因禍得福 看見希望
許多病友的人生是以確診做分水嶺,俊廷也是,28歲前的他,從來不知道罕見疾病是什麼,即使自己就是那位「萬分之一」的人,他也絲毫不覺。俊廷其實從小就發病了,身體不同部位不定期腫脹,如皮膚吹了顆小氣球,一點點脹痛和發熱感。俊廷每次發作都是單側,這次左腿腫,下次右手腫,醫院查不出原因,腫在手指就當作過敏、腳腫就當作痛風。
每回腫脹大約2到3天便會消去,對生活其實沒太大影響,一直認為自己是痛風的他,「從善如流」跟著爸爸吃痛風藥,現在想來都會搖頭偷笑。28歲時的發作很不尋常,頭一回腫在喉嚨,俊廷感覺喉頭好像被什麼卡著,呼吸越來越不順。急診醫師用內視鏡一看,發現他的呼吸道只剩下百分之十,緊急進手術室做氣切,情況危急到麻醉師還沒打麻醉,便已因呼吸不到而暈了過去。
幸好,手術成功,喉嚨腫脹消退,主治醫師仔細詢問俊廷的病史,發現俊廷的媽媽和妹妹都有類似症狀,媽媽甚至更嚴重,腫的部位遍及臉和全身。醫師推測是遺傳疾病的情況下,全家人抽血驗基因,最終確診是罹患「遺傳性血管性水腫」。一場急性發作讓他多年來無解的毛病終於知道如何醫治,無疑是上帝開的明亮之窗。
確診後,俊廷了解到此疾病與免疫功能有關,大學時常熬夜、壓力大,發作較為頻繁,每2~3個月便會發作一次。現在每個月回診拿口服藥,副作用輕微,久久發作時,腫脹程度也較以往輕微、持續的時間也較短。
走過人生關卡,俊廷始終保持正向、樂觀的心情,如今疾病控制得很好,現年35歲的他,成立遺傳性血管性水腫病友聯誼會,希望能找出更多同樣疾病的病友,幫助他們在診斷與治療的過程中能更加順利,同時俊廷正摯劃人生下一階段的藍圖呢!
認識罕見遺傳疾病系列(一一八)
遺傳性血管性水腫(Hereditary Angioedema, HAE)是一種補體缺陷的免疫不全疾病。補體由許多分子組合,是存在於人體血清、組織液及細胞膜表面具有活性的蛋白質,在人體中進行免疫調節抵禦外來微生物的侵襲。C1抑制蛋白(C1 esterase inhibitor,C1-INH)是補體的調節因子之一,當C1抑制蛋白的功能發生缺陷,影響人體免疫調節,使血管滲透性增加,造成發炎反應。HAE發生率為1/50,000,大多為體染色體顯性遺傳,第一與第二型HAE其中約有20-25%患者為新突變(de novo)。
主要臨床特徵為:反覆發作的皮下或黏膜下水腫,主因於過量血管外積液,阻塞血液或淋巴液的流動導致四肢、面部(眼瞼、嘴唇、舌頭)、生殖器、腸胃道或呼吸道(咽喉)腫脹。發作時皮膚會刺痛但不癢、或有邊緣性紅斑與局部硬腫。當有腸胃道腫脹會使患者出現噁心、嘔吐及劇烈腹痛的症狀;呼吸道(咽喉)腫脹造成疼痛、吞嚥困難和發音障礙,嚴重導致窒息而危及性命。此外抗組織胺藥物及皮質類固醇治療無法有效改善HAE症狀也為特徵之一。
通常在兒童時期出現症狀,於青春期階段惡化,終生持續,症狀嚴重程度因人而異,無法預測。沒有經過治療的患者,平均每1~2週會發作一次,約3~5天症狀才會緩解。HAE症狀發作的誘因可以為壓力、創傷、感染、雌激素及過度疲勞。HAE目前分為三種型別:
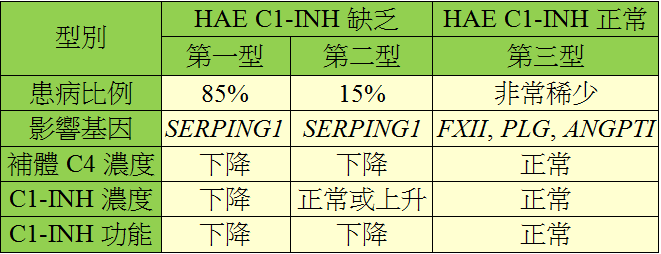
治療包括:新鮮冷凍血漿(fresh frozen plasma, FFP)、雄性激素(androgen)、抗纖溶劑(antifibrinolytic agents)、血漿製成C1抑制蛋白(plasma-derived C1 inhibitor)、基因重組C1抑制蛋白(recombinant human C1 inhibitor)、激肽系統調節劑、單株抗體等作為緩解HAE急性發作、短期或長期預防,目前多種新藥尚在研發中。
建議患者記錄每次發作情況,在正常生活狀況下盡量避免誘發因子,定期回診追蹤並配合治療,以改善生活品質。
遺傳性血管性水腫單張下載

