聯合報/2019.03.01/記者陳雨鑫
出生就有一張老臉、大耳、如蜘蛛腳般的手腳趾,這是極為罕見的疾病Galloway-Mowat症候群,台灣的案例幾乎活不過3歲,過去一直找不到主要的關鍵基因,台北馬偕醫院結合各科的專業,經年研究,終於發現屬於台灣患者致命基因KEOPS複合基因中的OSGEP(GMS3)缺陷,該研究也發表於知名罕見疾病期刊(Orphanet Journal of Rare Diseases)中。
Galloway-Mowat症候群是種罕見疾病,屬遺傳性疾病,且為隱性基因遺傳,在1968年被發現,台灣首名案例在1994年於台大醫院發表,第二名案例則在1999年於馬偕發現,至今已滿20年,目前全球已發表案例約有60多名,台灣就占10名。
台北馬偕醫院小兒腎臟主任蔡政道說,該罕病屬於遺傳性疾病,媽媽在懷孕期間,孩子的腦部發展及腎功能無法健全發展,孕婦通常會有羊水不足、胎兒腦部比一般胎兒小的現象,可藉由核磁照影(MRI)發現。
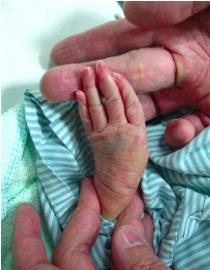
Galloway-Mowat症候群的孩子出生後,外觀與一般嬰兒有很大的差異,蔡政道說,通常會有大耳朵,且耳朵薄如水餃皮,五官下垂像極「阿公臉」、額頭凹陷、手指腳趾就像是蜘蛛腳一般細長、水腫等,嬰兒的活動力也極差,通常外觀都符合上述描述幾乎可以下診斷。
Galloway-Mowat症候群的孩子腎功能不佳,不斷在排蛋白尿,蛋白質大量流失,使孩子一出生就必須要透過藥物控制,同時腦部的灰質、白質突變,小腦嚴重萎縮,蔡政道說,全球的案例平均可以活到6歲左右,但台灣的孩子皆活不到3歲,最長的只有1年9個月;最短則是只有2個月。
由於國際研究的案例與台灣的症狀有所不同,馬偕醫院罕見疾病中心主任林炫沛與國外學者合作,針對國際上907位有腎病症候群的孩童,進行全基因定序檢查,其中有91位被診斷Galloway-Mowat症候群患者,其中在37位患者中找到KEOPS複合體基因缺陷,其中也包含台灣10名確診的患者,該研究也於2017年發表。
林炫沛說,Galloway-Mowat症候群患者有KEOPS複合體基因缺陷的消息令人振奮,但台灣患者的症狀與其他國家的患者有明顯的差異,他著手針對台灣的患者做一研究,針對台灣確診患者中,其中的6名患者做一研究,發現台灣的患者都是KEOPS複合體基因中的OSGEP(GMS3)缺陷,也證實有該基因缺陷的患者,症狀都相對嚴重,幾乎屬於致命基因。
追溯Galloway-Mowat症候群患者的家族,曾有一個家庭表述,過去曾有家人產子後,孩子不到幾個月就過世,且該名孩子也有類似的外觀特徵,林炫沛深信台灣還有非常多的患者未被發現。
他也呼籲,若家中曾有孩子因不明原因夭折,且有類似的外觀特徵,女性在懷孕期間得多觀察自己的羊水狀況,一旦有羊水過少的情形,建議可以加做核磁照影以及全基因定序,一旦確定孩子是Galloway-Mowat症候群,建議引產。
●Galloway-Mowat症候群小檔案
起源:1968年由加勒威(Galloway)醫師及莫沃特(Mowat)發現此病症,並以2人名字命名之。
基因:KEOPS複合基因缺陷。目前已知KEOPS複合基因有LAGE3(CGMS2)、OSGEP(CGMS3)、TP53RK(CGMS4)、TPRKB(CGMS5),台灣的個案皆是KEOPS複合基因中的OSGEP(CGMS3)缺陷,多屬嚴重類型。
基因缺陷比例:東南亞有0.0008的機率
疾病發生率:百萬分之一
病因:先天腦部發展異常造成小腦症合併腎病症候群
臨床症狀:精神不濟、大量蛋白尿、低白蛋白血症、水腫、高膽固醇、免疫力差
外觀特徵:大耳朵、額頭狹窄、小下巴、眼睛嘴巴下垂如老人、手指腳趾如蜘蛛腳
平均壽命:小於6歲死亡,台灣案例皆在3歲前死亡
案例:全球發表案例約60名、台灣占其中10名
資料來源/馬偕醫院罕見疾病中心主任林炫沛、馬偕醫院小兒腎臟科主任蔡政道
新聞出處:https://udn.com/news/story/7266/3672024 |