| 分類代碼: 0309 | ||||||||||||
| 疾病類別: 03 | ||||||||||||
| 疾病名稱: 亞硫酸鹽氧化酶缺乏 ( Sulfite Oxidase Deficiency ) | ||||||||||||
| 現階段政府公告之罕見疾病: 有 | ||||||||||||
| 是否已發行該疾病之宣導單張: 沒有 | ||||||||||||
|
ICD-9-CM診斷代碼:270.0
ICD-10-CM診斷代碼:E72.19 病因學: 微量元素鉬(Molybdenum)在體內會代謝成輔酶(cofactor),幫助亞硫酸鹽氧化酶(sulphite oxidase)、黃嘌呤氧化酶(xanthine oxidase)以及乙醛氧化酶(aldehyde oxidase)三種酵素作用。缺乏任一酵素可能會造成體內次黃嘌呤(Hypoxanthine)及黃嘌呤(xanthine)代謝成尿酸(uric acid)和亞硫酸鹽(Sulfite)轉變成硫酸鹽(Sulfate)的代謝過程受阻。(相關內容請參考鉬輔酶缺乏症) 亞硫酸鹽氧化酶缺乏(Sulfite oxidase deficiency),又稱為胱氨酸尿症(Sulfocysteinuria)。1967年,Mudd等醫師發現到一名患有嚴重神經退化及水晶體異位的嬰兒,在尿液中出現過高的亞硫酸鹽(Sulfite)及過低的硫酸鹽(Sulfate),而發現此一疾病。
由SUOX基因(Sulfite oxidase gene)所調控的亞硫酸鹽氧化酶(Sulfite oxidase),可使體內的亞硫酸鹽(Sulfite)轉化為硫酸鹽(Sulfate);一旦該基因有所缺損,導致酵素的缺乏,造成亞硫酸鹽的過度堆積,進而損害腦、神經系統等重要器官,甚至造成死亡。
此疾病為體染色體隱性遺傳模式,在同一個家族中,可能出現多位罹病的兄弟姊妹;Mudd et al. (1967)的研究中發現,該嬰兒也有疑似因相同症狀而過世的手足。Vianey-Liaud et al. (1988)更在一近親通婚的阿爾及利亞家庭中,發現五位前後皆死於此一疾病的兄弟姊妹。
遺傳模式 為一體染色體隱性遺傳,若父母為帶因者,每一胎有25%的機率生下患者,50%的機率是跟父母一樣的帶因者,25%的機率正常。 發生率:
此症發生率極低,截至目前約有50位案例被發表出來,尚未有疾病發生率的推估。 臨床症狀: 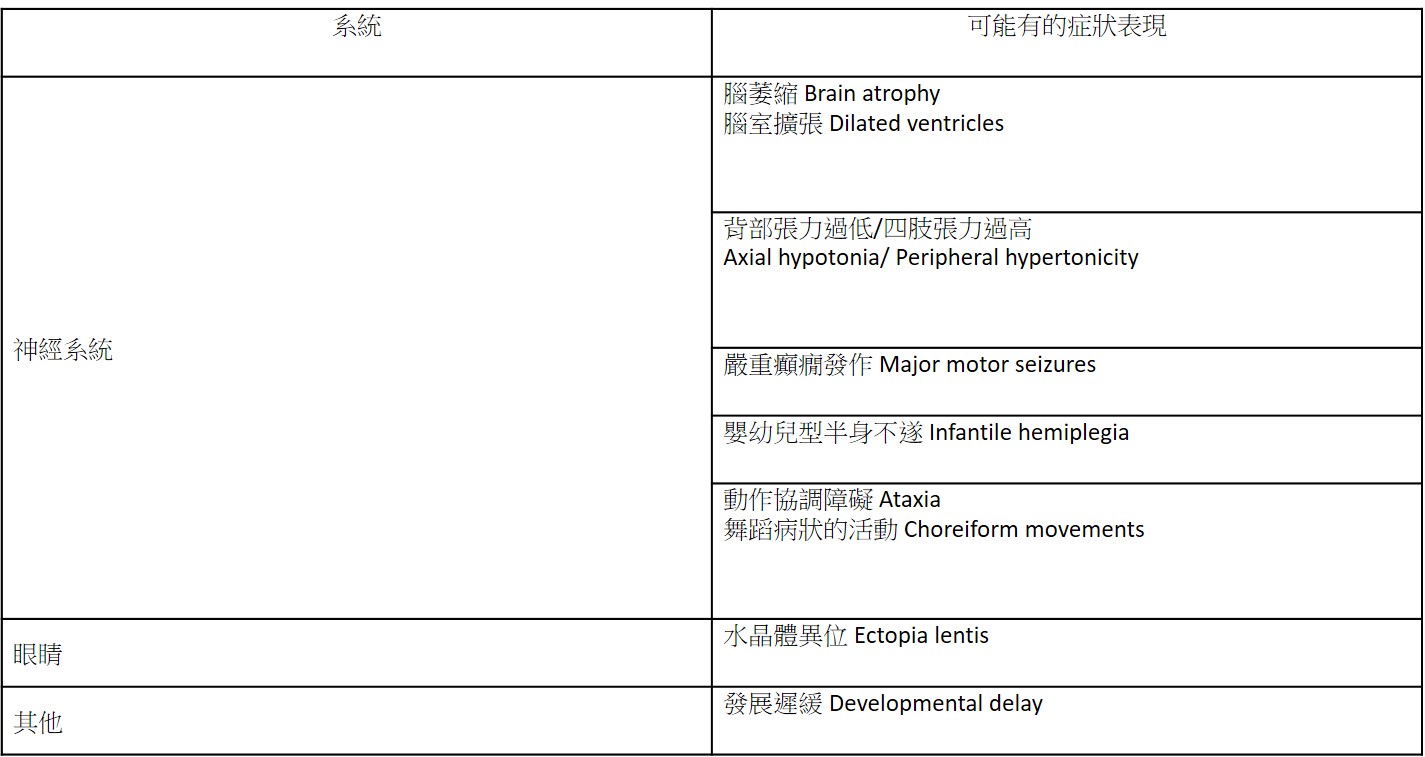
診斷: 1.快速篩檢: 新鮮尿液亞硫酸鹽測試(Sulfite test)試紙測驗陽性
2.血液及尿液檢查:
3.影像檢查:腦部電腦斷層、核磁共振檢查等,評估患者腦部病變的程度。 4.基因檢查:抽血經分子生物技術,檢測是否有SUOX的基因缺陷。 治療: 可嘗試予以甜菜鹼(Betaine),增加同半胱氨酸(homocysteine)退回蛋胺酸(methionine,又稱甲硫丁胺酸)的重甲基化(remethylation)的能力,以減少半胱氨酸(cysteine),期望能因此降低過量堆積的亞硫酸鹽(Sulfite)。在飲食的部分,曾經有人嘗試使用低蛋白限制甲硫胺酸飲食治療,但效果未明。 另外,給予高劑量的維他命B1(thiamine)可以補充因為過量亞硫酸鹽(Sulfite)造成維他命B1的消耗。目前尚沒有任一藥物可以治癒這個疾病,主要仍是以症狀治療如癲癇和痙攣的控制為主。
預後: 1.OMIM- https://www.omim.org/entry/272300 2.Medscape- https://emedicine.medscape.com/article/949303 3.Sulfite oxidase deficiency in man: demonstration of the enzymatic defect. Mudd SH, Irreverre F, Laster L. Science. 1967 jun 23;156(3782):1599-602. 4.A new case of isolated sulphite oxidase deficiency with rapid fatal outcome. Vianey-Liaud C, Desjacques P, Gaulme J, Dorche C, Vanlieferinghen P, Dechelotte P, Divry P. J Inherit Metab Dis. 1988;11(4):425-6. No abstract available. 2015年委託台大醫院基因醫學部 審稿 |
