| 分類代碼: 1628 |
| 疾病類別: 16 |
| 疾病名稱: Ayme-Gripp症候群 ( Ayme-Gripp syndrome ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 有 |
|
ICD-9-CM診斷代碼: ICD-10-CM診斷代碼:Q87.89 疾病簡介 Aymé-Gripp症候群是合併以下三個症狀的疾病,包括雙側早期白內障(cataract)、感覺神經性聽力喪失(sensorineural hearing loss)及獨特的臉部特徵伴隨神經發育異常(characteristic facial features in combination with neurodevelopmental abnormalities)。患者的臉部看起來像唐氏症患者的臉,包括短頭畸形、面部扁平、鼻子短、面部扁平、鼻子短、長人中、嘴巴狹窄、低位且向後旋轉的耳朵。 其他症狀包括: 先天性聽力喪失、身材矮小、癲癇、非特異性腦部異常、骨骼關節問題等。目前被報導過的個案都有發育遲緩,而每位患者認知障礙的程度不一。其他比較少見的症狀包括:嬰兒時期有心包膜積水、腸胃道和內分泌問題、外胚層發育異常、牙齒症狀等。 衛福部國健署於2020年公告將此症新增至罕見疾病名單內。 病因學 此症是由MAF基因發生致病變異所致。此基因製造出的蛋白質在水晶體和眼睛發育的過程中扮演很重要的角色。且在胚胎發育過程中也控制著多個生理過程,包括軟骨細胞和皮膚細胞的分化。 發生率 未知。此疾病非常罕見,目前文獻上只有19個家庭中的21位個案被報導過。外顯率雖為100%,但即使在同家族中,嚴重程度上也會有明顯差異。 遺傳模式 此症為體染色體顯性遺傳。 目前報導過的案例有90%皆為自身基因產生新的突變引起的單純性病例(即家庭中唯一的患者)。Aymé-Gripp症候群的患者的每個孩子都有50%的機會遺傳到相同的致病變異。而其兄弟姊妹罹病之機率,則須根據父母是否帶有MAF致病變異決定,若父母有,則為50%,若父母未檢驗出,仍因父母染色體鑲嵌現象之可能性,風險估計為1%。若家族中確診患者已找到致病的突變,則可以對高風險的孕婦進行產前診斷。 臨床表徵 患者經常有雙側早期白內障、感覺神經聽力喪失和與唐氏症相似的面部,這三種特徵。 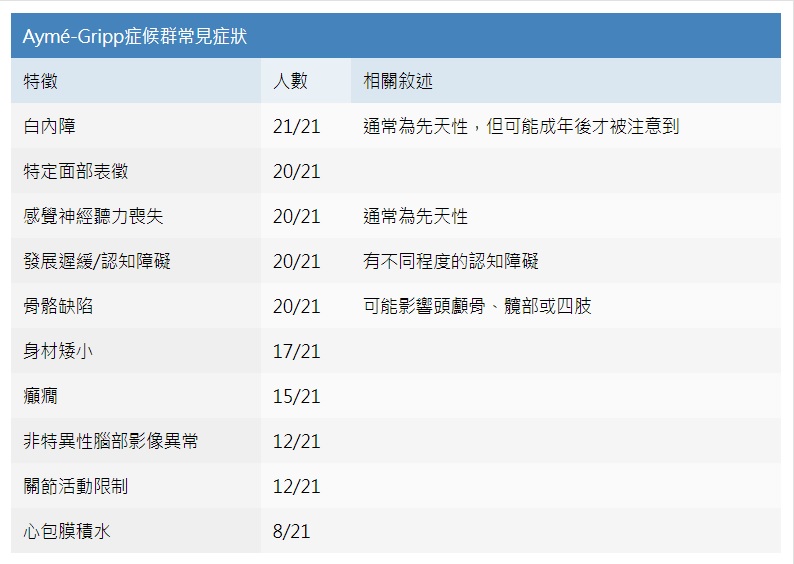 ◎特定的面部特徵: 短頭畸形(brachycephaly)〈約80%〉、面部輪廓扁平(flat facial appearance)、中臉凹陷、短鼻(short nose)、長人中(long philtrum)、薄上唇及嘴巴窄小(narrow mouth)、牙齒異常(小而畸形的牙齒)、低位且後旋的耳朵。其他特徵含眼距較寬、寬且稀疏的眉毛、前髮際線高。 ◎聽力方面: 通常是雙側的聽力損失,大部分為先天性或早發性,且多在兒童早期被診斷。聽力損失的程度從輕度到重度不等,少數患者需要助聽器或人工耳蝸。在21名案例中,只有一名成年的母親聽力為正常。 ◎眼科表現: 幾乎所有患者都有先天性或早發性白內障。白內障最早可在嬰兒期發現,但有些直到青春期甚至成年才發現。 ◎生長表現: 多數患者身材介於矮小到中等之間,身高低於平均年齡1.25-2個標準差。 通常在出生前的身高發育正常,出生身高在正常範圍內,出生後身高成長逐漸落後;但體重和頭圍的發育通常都在正常範圍內。 ◎認知發展: 文獻報告患者會有語言和運動能力的遲緩。有些患者也被診斷有自閉症光譜疾病。除了一名帶有變異的父母無認知學習障礙,順利取得學士學位外,其餘所有人皆有發展遲緩的現象,但不同患者認知障礙程度差異很大。如一對母子只有輕微的學習障礙,且皆完成了中學教育;但另一個29歲的個案有語言障礙,僅能使用手語進行交流。 ◎神經系統表現: 在21名患者中,有15名罹患癲癇,其中5名患有熱性痙攣。這些患者的發病年齡介於六個月至十二歲之間。有半數以上的患者曾接受腦部影像檢查,發現非特異性的腦部異常,包括腦室肥大(ventriculomegaly)、小腦扁桃體下疝畸形(Chiari I malformation)、阻塞性水腦症(obstructive hydrocephalus)、空蝶鞍症候群(empty sella)以及腦萎縮(erebral atrophy)。其中有一位患者有基底核鈣化現象。 ◎關節和骨骼表現: 大多數患者都有關節問題,症狀表現差異很大,從先天尺橈近端關節粘連伴隨前臂短小到輕微的活動受限。其中,先天尺橈近端關節粘連、橈骨頭半脫位及前臂短小為最常見的關節表現。 另外還有手指(腳趾)彎斜、 屈曲指(一個或多個指頭固定於屈曲狀態的變形) 、短指(趾)症、漏斗胸、脊椎側彎、骨齡延遲、腕/跗骨缺損、第四蹠骨短小以及髖關節壞死等;顱縫早閉屬於相當罕見的特徵,需手術修復。 ◎心臟表現: 在新生兒或嬰兒期,發現21個人中至少有8個人罹患心包膜炎或心包膜積水。另外各有一名案例診斷心房中膈缺損及開放性動脈導管。 ◎外胚層表現: ·女性可能有雙側乳腺發育不全。 ·頭髮:不論是兒童或是成人都有可能頭髮稀疏。某些成人患者發現輕至中度過早掉髮的現象。 ·指甲:指甲營養不良或瓣狀甲。 ·牙齒異常:約有不到半數的患者會有牙齒較小、畸形牙齒或少齒的狀況。 ◎內分泌表現: 只有3個案例診斷有甲狀腺功能低下,這些患者非先天性甲狀腺功能低下,而是在兒童期被診斷,並對甲狀腺激素替代療法反應佳。其他內分泌異常很少被報導。 ◎其他罕見表現: ·裂孔疝氣及橫膈膜疝氣 ·短暫性血尿 ·蛋白尿或尿中微白蛋白 ·腎絲球腎炎 ·腎病症候群(類固醇治療後未復發) 診斷方式 除上述三個主要特徵外,可以再藉由分子基因檢測(molecular genetic testing)確認,此症在MAF基因上會帶有致病性變異。目前尚未完全建立正式臨床診斷準則。如個案具有下列主要及次要的臨床特徵與影像學發現,則應該懷疑患有Aymé-Gripp syndrome的可能。 ◎ 主要臨床特徵: 白內障、感覺神經性聽力喪失、獨特面部特徵、發展遲緩/智能障礙、身材矮小。 ◎次要臨床特徵: 癲癇(包括從熱痙攣到癲癇症)、先天性尺橈近端關節粘連(congenital radioulnar synostosis)、心包膜積水、乳腺發育不全、皮膚外胚層異常(稀疏的頭皮頭髮,營養不良的指甲)。 ◎影像學: 腦部影像異常(腦室肥大、腦積水、腦萎縮)、先天尺橈近端關節粘連、橈骨頭半脫位、髖關節軟骨壞死(年輕成年人)、腕/跗長骨缺損。 ◎分子基因檢測 由於Aymé-Gripp syndrome的表現型很廣泛多樣,如果發現個案符合Aymé-Gripp syndrome的獨特臨床特徵時,建議可以針對MAF基因進行檢測。 治療 此症的治療方向主要是對發生的臨床症狀進行處理,理想的治療包含多個不同學科的專業處置,包括眼科、耳鼻喉科、骨科、內分泌科、心臟科、神經科、 發展和行為專家、語言治療師、職能治療師及物理治療師,具體治療取決於患者的臨床症狀需求。 |
