| 分類代碼: 0117 |
| 疾病類別: 01 |
| 疾病名稱: 三甲基巴豆醯輔酶A羧化酵素缺乏症 ( 3-Methylcrotony-CoA Carboxylase Deficiency, 3-MCC Deficiency ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 有 |
|
ICD-9-CM診斷代碼:270.9 ICD-10-CM診斷代碼:E71.19 前言: 3-甲基巴豆醯輔酵素羧化酵素缺乏症又名三甲基巴豆醯基甘胺酸尿症(3-methylcrotonylglycinuria),是一種罕見先天性代謝異常疾病。3-甲基巴豆醯輔酵素羧化酵素為代謝白胺酸所需之酵素,可以在生物素(biotin)的幫助下,將3-methylcrotonyl-CoA轉換成3-methylglutaconyl-CoA。缺乏此酵素將造成體內無法代謝白胺酸(leucine),導致三甲基巴豆醯輔酶甘胺酸(3-methylcrotonylglycine)的累積(如圖一)。 遺傳模式: 國外的發生率約為1/50,000,自從大規模使用串連質譜儀作為新生兒篩檢的工具後,全世界包括英,美,澳洲,與台灣皆報告這是所有篩檢疾病中最常見的有機酸血症,其發生率在台灣地區約為1/11,000。 此病症致病基因是位在第3對染色體長臂(3q25-q27) 上的MCCC1或稱MCCA(Methylcrotonyl-CoA carboxylase alpha chain)及第5對染色體長臂(5q12-13)上的 MCCC2或稱MCCB(Methylcrotonyl-CoA carboxylase beta chain)基因。 此症的遺傳方式大多為體染色體隱性遺傳,即父母雙方皆為帶因下,每一次懷孕產下此疾病機率為25%,產下帶因者機率為50%。但是體染色體顯性遺傳亦有人報導過。病患可能有父母或兄弟姐妹是具有3-甲基巴豆醯輔酵素羧化酵素缺乏症而無症狀或家庭成員可能有肌肉低張力或高張力的家族史。 症狀: 患者的臨床症狀差異大,可從症狀嚴重到無症狀。大多數的患者可正常生長與發育且並無症狀。少數報告案例指出患者於新生兒期的發展正常且無症狀,直到他們6個月至3歲之間因急性發作而被診斷(也有患者在成年發病),症狀包括餵食困難、嘔吐、呼吸暫停、嗜睡、低張力、抽搐、反射反應過強、低血糖、輕微的代謝性酸中毒、高血氨、肝功能異常、酮尿、血漿中游離態肉毒鹼值低、肌肉萎縮、抽痙以及皮膚炎或脫毛;急性發作通常在發燒生病、感染或長期飢餓後發生,而病患易在第一次嚴重代謝異常中會發生嚴重代償機能減退或死亡。有些患者在新生兒期會生長遲緩或發展遲緩。 無症狀的女性患者有可能將三甲基巴豆醯輔酶甘胺酸前驅物(3-Hydroxyisovaleryic acid)的代謝產物,經由胎盤傳輸給胎兒,導致新生兒的串聯質譜儀篩檢值出現偽陽性,而實際上新生兒並未罹病,若有上述情形發生,應再對新生兒的母親進行檢驗。 診斷: 1.通常可先經由新生兒篩檢方法中,實驗室使用串聯質譜儀分析血液中C5-OH數值。當數值為陽性時,尋求兒科新陳代謝科專家或遺傳學家諮商並做確認和診斷,由尿液中有機酸分析做診斷的確立。 2.皮膚纖維母細胞或白血球檢測3-甲基巴豆醯輔酵素羧化酵素定量。 3.直接進行MCCA/MCCB 基因突變分析。 治療: 急性發作時應積極矯正脫水,平衡電解質,改善代謝性酸中毒,並以高濃度葡萄糖持續灌注。另外,可給予病患甘胺酸(glycine)藥物治療以增加三甲基巴豆醯輔酶甘胺酸(3-methylcrotonoyl glycine)在代謝危機期間的排泄。 長期治療以控制飲食中白胺酸為主。生物素的給予對病人的幫助未有定論。若病人血液中游離態肉毒鹼(carnitine)降低時應該給予50~100 毫克/公斤/天 的carnitine。另外要避免長期飢餓,以免引起身體代謝走向分解蛋白質的路徑,而產生毒素堆積。 3-甲基巴豆醯輔酵素羧化酵素缺乏症若經早期診斷及治療,病患大多可以正常的生長和發展。 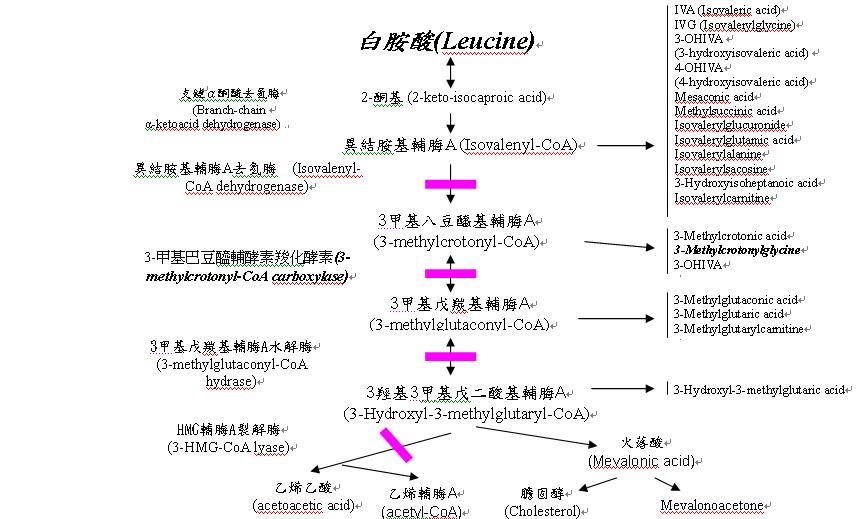 Reference: 1. (3MCC) 3-METHYLCROTONYL-CoA CARBOXYLASE DEFICIENCY: http://www.idph.state.ia.us/genetics/common/pdf/3mcc.pdf 2. 3-@METHYLCROTONYL-CoA CARBOXYLASE 1 DEFICIENCY:OMIM: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=210200 3. 3-Methylcrotonyl-CoA Carboxylase Deficiency (3MCC) http://www.savebabies.org/diseasedescriptions/3MCC.php 4. http://www.pediatrix.com/body_screening_menu.cfm?id=1569 5. http://www.genes-at-taiwan.com.tw/genehelp/Default.asp?kid=M 罕見疾病基金會南部辦事處 邱幸靜主任 編譯 台大醫院基因醫學部 李妮鍾醫師 審稿 2015年委託台大醫院基因醫學部 審稿 其他有關此項疾病之介紹請詳見「罕見遺傳疾病一點通」網頁及本會「認識罕見遺傳疾病系列」及「罕見疾病叢書之醫療飲食手冊」網頁(1)、(2) |
