ICD-9-CM診斷代碼:756.51
ICD-10-CM診斷代碼:Q78.0
成骨不全症(Osteogenesis Imperfecta)指的是一種因先天遺傳性缺陷,而引起膠原纖維病變,造成骨骼強度耐受力變差而容易脆弱骨折的疾病。臨床上的表現差異很大,它可以由較輕微的骨質疏鬆表現,至頻繁骨折,甚至在子宮內胎兒階段即產生骨折,最嚴重的情況在嬰兒出生不久即夭折,甚至造成死產。男性女性罹病的機率大約相同,統計上每10萬人中會有6〜7位患者。膠原纖維病變除了造成骨頭脆折,還會造成其他軟組織症狀,包含:身材矮小、藍灰色鞏膜、牙本質發育不良、聽力損傷、關節韌帶鬆弛。根據遺傳基因或骨骼脆度及骨骼系統外的表徵,目前依疾病特徵及嚴重程度分成5種不同的疾病分型。
第1型為體染色體顯性遺傳造成的正常膠原纖維形成不足,嚴重程度從輕微到中度都有可能,但大部分症狀較輕微的骨折。第1型外觀上鞏膜有明顯的藍眼珠(Blue sclera),少數個案在嬰兒出生時即會發現股骨彎曲。第一次骨折可能發生在出生時或換尿布時。更常見的是在嬰兒開始走路,骨折通常以每年幾次的頻率發生,而在50歲以後的男性及停經女性會更頻繁發生。雖然全身可能有幾處骨折到100多處不等,但骨折通常會正常癒合,不會導致畸形。大多數受影響的個體身高正常或接近正常,但通常比其他家庭成員矮,並且比平均父母身高預測的矮。關節過度活動容易導致一些輕微的合併症,主要是早發性退行性關節炎。牙本質發育不全在第1型個案不常見但仍要注意。大約50%第1型個案的成年人會出現進行性聽力損失,最初是傳導性聽力損失,但通常會出現額外的神經性聽力損失成分。
第2型的成骨不全症為最嚴重的,通常在子宮內即有多發性骨折而胎內死亡或自然流產,在出生前後的時期(Perinatal)常造成併發症死亡,甚至死產。異常特徵在出生時很明顯。體重和長度對於胎齡來說都很小,頭骨對於身體大小來說很大並且觸診柔軟。鞏膜呈深藍色,摸的到肋骨上的骨痂(callus),四肢短而彎曲,臀部通常彎曲並外展成蛙腿姿勢。超過60%的嬰兒在第一天死亡; 80%在第一周內死亡;超過一年的存活率非常罕見,死亡通常是由於與小胸相關的肺功能不全、肋骨骨折或由於肋骨不穩定而導致的連枷胸,需要特別加強呼吸道的支持通氣。它基因的異常,可能包括體染色體顯性或隱性遺傳,也可能是鑲嵌化(Mosaic)的異常,即有部分細胞正常,有部分異常;基因突變的結果導致皮質骨中的膠原蛋白極度缺乏,因此症狀特別嚴重。
第3型的成骨不全症的遺傳異常,可能為體染色體顯性遺傳,或者是體染色體隱性遺傳,導致第一型膠原纖維的結構異常。與其他型不同的是,第3型會有漸進式的骨骼變形。新生兒時期的骨折很常見,部分個案因嚴重肋骨骨折導致在生命的最初幾週或幾個月內因肺衰竭而死亡。存活下來的兒童因嚴重骨骼脆弱和明顯的骨骼畸形,通常使用輪椅或其他幫助來移動。即使沒有明顯的骨折,全身也會有多達 200 處骨折和進行性畸形。進行性骨骼變形通常很難在骨科上進行處理,即使使用髓內棒放置也是如此。生長極其遲緩,有些人的成年身高不到一米。鞏膜在嬰兒期是藍色的,但隨著年齡的增長而變淺。聽力損失在青少年時期會發生。頭臉部因進行性骨骼變形及牙本質發育不良而有腦室擴大、大頭、三角型臉,但部分人仍可保持正常的牙齒及面容。智力是正常的,但有文獻報導COL1A2基因的第49號外顯子的致病變異會增加顱內出血的風險而影響智力。另外,第3型病患須注意顱底壓迫造成的腦幹受壓、阻塞性腦積水或脊髓空洞症,平時應避免頸部屈曲時的壓迫症狀,如:後顱骨疼痛、第2節頸椎神經感覺障礙、第4和第5指刺痛以及前臂內側麻木。嚴重時會有三叉神經痛、四肢功能喪失或感覺異常的頭痛及發生睡眠呼吸暫停和死亡。
第4型為體染色體顯性遺傳或者是體染色體隱性遺傳,導致前膠原纖維(pro-alpha collagen)的結構異常。鞏膜在出生時通常為淺藍色或灰色,但很快變淺至接近正常。其他特徵是輕度身材矮小、常見牙本質發育異常、成人發病的聽力損失、脊椎側彎、嚴重程度從輕度到中度不等的骨折及骨骼變形。
第5型的臨床表現和第4型很相似,主要特徵是肥厚的骨痂、緻密的幹骺端(metaphyseal band)、前臂骨間膜(interosseus membrane)骨化。第5型為IFITM5基因的體染色體顯性遺傳,導致骨礦化(bone mineralization)的異常,因此骨頭的病理組織的特徵是不規則排列或網狀排列的骨板(lamella)。
相關基因及遺傳模式整理如下[3]:
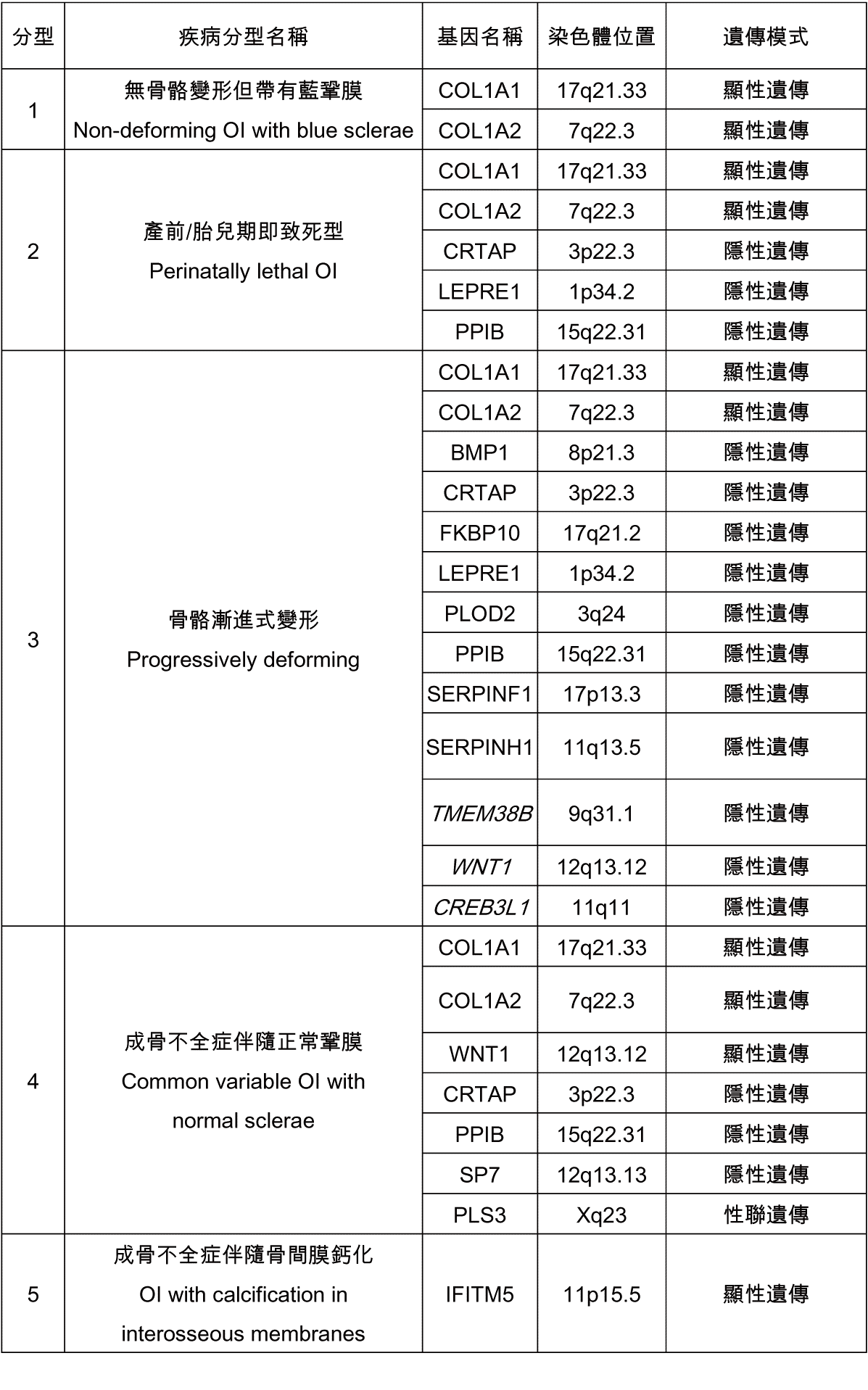
一般而言,它可以在同一家族出現較一致的外觀表現,但也可能在同一家族有不同的外表表現;不同家族而表現出類似外觀者也有可能具不同的膠原缺陷。臨床照顧上須多專家團隊協助,包含臨床遺傳學、骨科、復健、兒科牙科、耳鼻喉科和身心科,家人小心的照顧也是很重要的。主要治療方法包括:根據嚴重程度支撐四肢;用於穩定鬆弛關節的矯形器;物理和職能治療,以最大限度地提高骨骼穩定性、改善活動能力、防止攣縮、防止頭部和脊柱畸形,並改善肌肉力量和疼痛處理。防止骨折,可以氣墊褲或使用骨髓內固定釘加強骨骼應變力。骨折的治療方法:盡可能短的固定時間;小而輕的骨固定器;移除石膏後立即進行物理治療。雙磷酸鹽無法降低骨折頻率,但可以改變骨質密度、成人身高及疼痛感。
以上資料轉錄自本會成立特刊 林口長庚醫院骨科 李宗料醫師提供
2017年委託台大醫院基因醫學部 審稿
2021年歐宗穎/李妮鍾醫師 審閱更新
參考資料:
1. Osteogenesis imperfecta:clinical diagnosis,nomenclature and severity assessment. Am J Med Genet A. 2014 Jun;164A(6):1470—81. Doi:10.1002/ajmg.a.36545. Epub 2014 Apr 8.
2. Genereviews:COL1A1/2Osteogenesis Imperfecta
3. Van Dijk FS,Sillence DO. Osteogenesis imperfect:clinical diagnosis,nomenclature and severity assessment. Am J Med Genet A. 2014 Jun;164A(6):1470—81. Doi:10.1002/ajmg.a.36545. Epub 2014 Apr 8. Erratum in:Am J Med Genet A. 2015 May;167A(5):1178. PMID:24715559; PMCID:PMC4314691.
其他有關此項疾病之介紹,請詳見「罕見遺傳疾病一點通」網頁、本會「認識罕見遺傳疾病系列」網頁、本會「照護手冊」網頁、本會繪本「螢火蟲故事島第一季-玻璃娃娃的天空」
|
