| 分類代碼: 0904 |
| 疾病類別: 09 |
| 疾病名稱: Nemaline線狀肌肉病變 ( Nemaline Rod Myopathy ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 有 |
|
ICD-9-CM診斷代碼:359.0 ICD-10-CM診斷代碼:G71.21 病因學
是一種少見的先天性肌肉病變,特徵是全身肌肉無力,特別是在臉部、頸部、軀幹跟近端肌肉的部分,大部分患者在出生時症狀最為嚴重,隨著年齡增長患者的肌肉無力多呈現穩定,動作發展還是可能會緩慢進步。有許多基因會導致此症的發生,這些基因跟維持骨骼肌的正常結構與功能有關,因此一旦這些基因發生致病突變,便會對肌肉收縮的能力造成影響。此症最常見的致病基因是NEB跟ACTA1基因,NEB基因突變約占所有患者的50%左右,症狀通常在出生後沒多久或童年早期發病;ACTA1基因突變則佔15%~25%左右,病情嚴重程度跟發病年齡差異很大。
患者可能會有吞嚥困難、足部畸形、脊椎側彎跟關節畸形的症狀。雖然有部分患孩學會走路的時間較慢,但大部分患者都可以行走。在嚴重的情況下,呼吸功能會受到影響而造成呼吸困難。
發生率
此症發生率目前約為5萬分之一。
遺傳模式
此症大部分為體染色體隱性遺傳模式,代表若父母親雙方同為此一缺陷基因帶因者,不分性別,每一胎則有1/4的機率會遺傳此症;但有少部分為體染色體顯性遺傳,代表若雙親其中一位患有此症,其子代則不分性別,每一胎有50%的再發率。不過大部分的患者都是在沒有家族史的自發性突變患者。
而遺傳模式會依據不同的致病基因而有所不同,如下表所示:
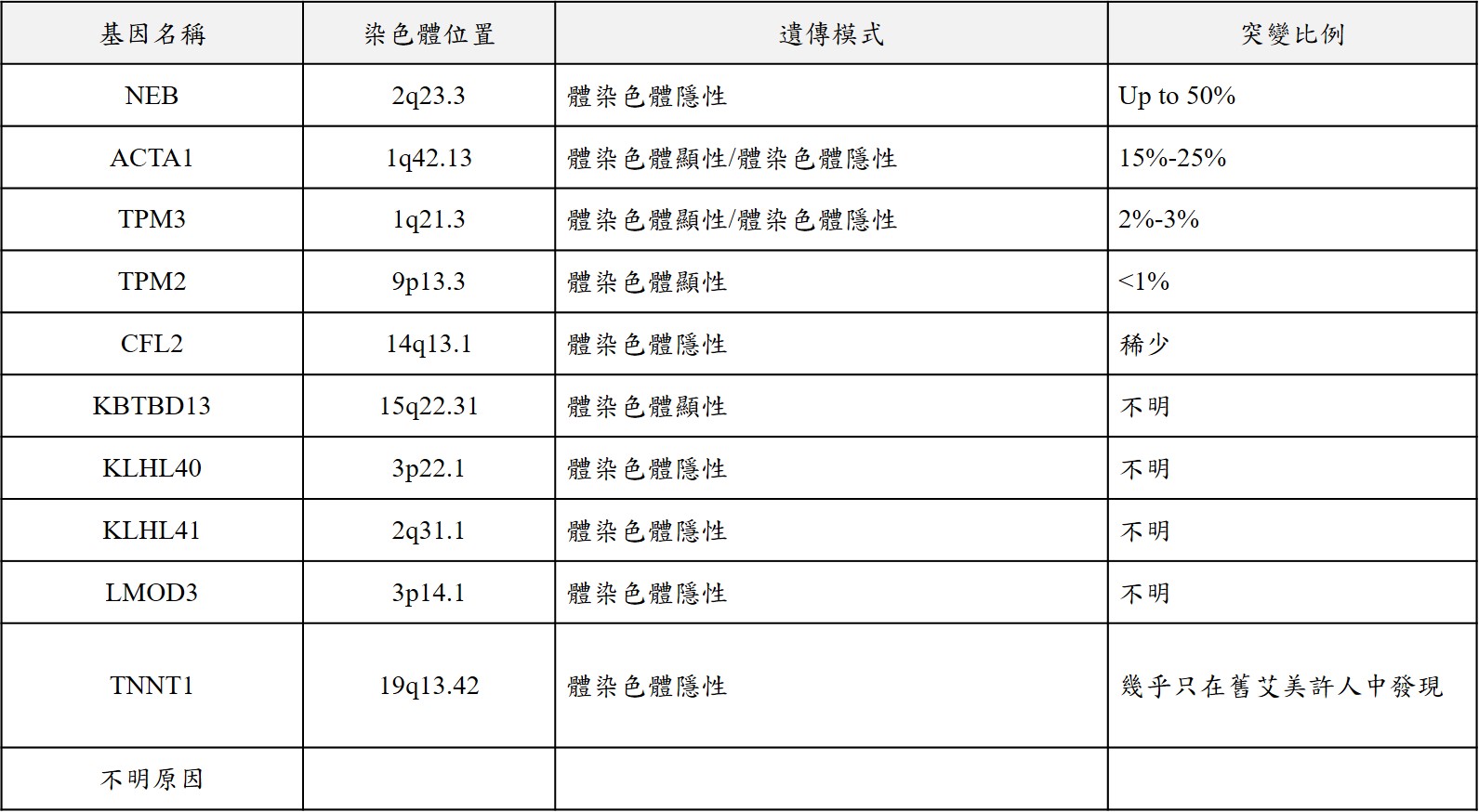 臨床症狀
此症的主要特徵是肌無力、肌張力低下和肌腱反射能力下降或消失,肌無力的狀況主要發生在面部、頸部跟近端肌肉,大肌肉動作發展較差,但大多數患者其他發育狀況是正常的。
嬰兒時期可能出現動作發育遲緩、生長遲滯及全身無力現象,全身低張力、呼吸衰竭、餵食困難。初期症狀為走路時易跌倒、爬樓梯困難。非進行性肌無力併肌萎縮、骨骼異形、瘦長臉頰、高口蓋、脊柱側彎、弓形足。神經檢查可能發現顏面肌肉與四肢近端肌無力,肌腱反射消失等症狀。
診斷
此類之先天性肌病最明顯的特徵在於肌肉的特異病理變化,可以觀察到肌肉組織中的肌肉結構的異常變化。此症肌肉切片的特徵是可發現明顯的線狀體(nemaline rod)存在。診斷上也需同時合併臨床症狀與肌肉病理檢查來加以確認。血清肌胺酸肌酵素(creatine, CK)則多為正常或輕微上升。
治療
此症主要還是依症狀治療為主,應由各領域的專家共同進行照護,治療及預防併發症的發生,如:呼吸道與肺部感染、呼吸功能不全、胃食道逆流、脊椎側彎和關節攣縮等。
目前有研究指出,使用左旋酪胺酸(L-tyrosine)可以改善患者的肌肉強度及清除口腔分泌物的速率。有部分口服左旋酪胺酸患者認為自身的狀況有所改善,特別是改善延髓功能和運動耐受性。在ACTA1突變的實驗小鼠中,也有發現左旋酪胺酸可以改善肌肉強度。因此左旋酪胺酸在未來可能可以作為治療此症的潛在藥品。
預後
建議對於每一個被診斷為先天性肌病患者,應接受整合性醫療照護,長期追蹤肺功能的變化,並接受復健治療以避免骨骼及關節之變形,以提高其生活品質。
2017年委託台大醫院基因醫學部 審稿 2022年翁妏謹醫師審閱更新 以上資料轉錄自「罕見遺傳疾病一點通」網頁 其他有關此項疾病之介紹,請詳見本會「認識罕見遺傳疾病系列」網頁 |
