| 分類代碼: 0317 |
| 疾病類別: 03 |
| 疾病名稱: 先天性高乳酸血症 ( Congenital Hyperlactic Acidemia ) |
| 現階段政府公告之罕見疾病: 沒有 |
| 是否已發行該疾病之宣導單張: 沒有 |
|
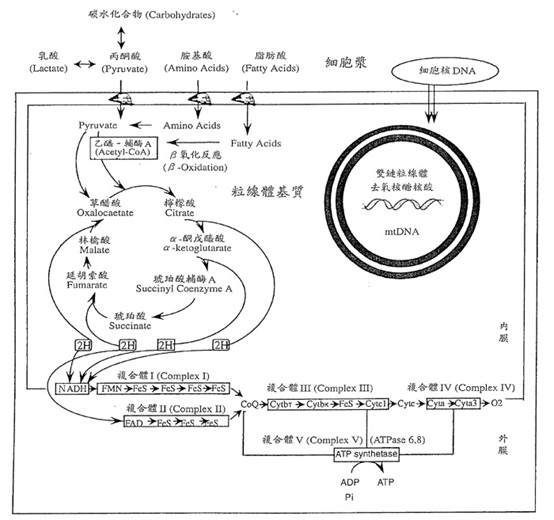
我們身體裡的細胞會通過醣解作用(glycolysis)分解葡糖糖和肝醣以獲得能量,而丙酮酸(pyruvate)為醣解作用之最終產物。一般情況下,生成的丙酮酸會通過粒線體內外膜進入粒線體基質,和乙醯輔酶結合後進入克氏循環(Kreb’s cycle又稱TCA cycle)進行代謝,透過一連串的氧化磷酸化的過程產生能量。但當氧氣缺乏或是進入克氏循環代謝途徑受影響時,丙酮酸則會經由乳酸去氫酶(lactate dehydrogenase, LDH)的作用轉換成乳酸(lactate)且同時快速產生能量給細胞使用。進而導致血液中乳酸值升高造成高乳酸血症(lactic acidosis)。
造成高乳酸血症的原因可分為兩大類,一類為非酵素缺乏的情況,可能因缺氧、藥物、抽搐等引起;另一類為酵素缺失所引起,包括:醣類、有機酸、維生素、脂肪酸及粒線體代謝異常。而先天性高乳酸血症是指因酵素缺失所引起的這群疾病。
先天性高乳酸血症是一群罕見代謝疾病的共同表現。其發生可以是由於細胞核的DNA(nDNA)或是粒線體DNA(mtDNA)產生一個或是多個遺傳或偶發的突變導致的。
 (圖片來源:李明亮 (2004).代謝性疾病:台灣經驗,合記圖書出版社,台灣) 遺傳模式: 1. 母系遺傳,是由於mtDNA點突變而致病 2. 偶發性,由於mtDNA缺失或重複而致病 3. 性聯遺傳、體隱性或體顯性遺傳。若因nDNA缺失所導致的,上述3種遺傳模式皆有可能。 發生率: 實際上的發生率不是很清楚,不過有一個預估統計顯示,美國每年每1000位活產新生兒中約有250-300位為先天性高乳酸血症的孩子。男生和女生的比例是一樣的。 臨床症狀: 因酵素缺乏導致的先天性高乳酸血症會因嚴重程度不同,影響的器官數目不同,因此出現不同的症狀。有些人可能血液、腦脊髓液和尿液中乳酸數值持續升高,而其他人可能只在壓力或其他外來原因,如感染、癲癇或哮喘發作引起時偶爾增加乳酸。
在那些嚴重酵素缺陷的兒童,先天性高乳酸血症的臨床表現可能在出生後的幾小時或幾天內就出現,症狀包含肌張力下降(低張力)、嗜睡、嘔吐和呼吸過快等。隨著時間推移而出現發展遲緩、智能障礙、運動異常、行為問題、甚至出現多重器官受到影響的表現,例如: 糖尿病、早期聽力喪失、失明、腎小管酸中毒、腎功能異常或是心臟衰竭等等。對於一些由於粒線體DNA突變引起的相對較輕微的個案,先天性高乳酸血症的併發症可能要到青春期或成年期才會出現。
以下是與此症相關之疾病簡介: (1) 丙酮酸去氫酶複合體缺乏(pyruvate dehydrogenase complex deficiency): 由於此酵素缺乏所導致的先天性高乳酸症約佔10-15%,致病基因是位於X染色體上的PDHA1基因,為X染色體性聯遺傳,臨床上患者男女都有,此型PDC缺乏症被歸為X-linked共顯性,對於女性患者具有多變性的臨床表徵,大多數的PDHA1基因突變是偶發性的。其他型更為罕見。
丙酮酸去氫酶複合體缺乏可能造成高乳酸血症,導致新生兒輕微發展遲緩,或是孩童、青少年的運動方面受損,乳酸與丙酮酸值會一起增加,血中兩者比值則為正常。腦脊髓液中的乳酸濃度較高於血中乳酸濃度。
丙酮酸羧化酶缺乏是由於nDNA基因缺陷所導致,為體隱性遺傳。此症分為三型,從輕微的至嚴重的高乳酸血症、神經發展遲緩、智力障礙及早夭都有可能發生。 (3) 延胡索酸酶缺乏(Fumarase deficiency): 粒線體的延胡索酸酶(Fumarase)是參與克氏循環(Krebs cycle)中的酵素之一,若有缺失的話可能造成先天性高乳酸血症。延胡索酸酶缺乏為體隱性遺傳。主要的症狀是腦部神經系統受到影響,小腦、腦部構造異常,生長、發展遲緩、肌張力不足及尿中延胡索酸增加;有些患者會有外觀異常,如:前額突出、眼距較寬、小下巴等。其它克氏循環酵素的缺陷也可能造成高乳酸血症,包括:烏頭酸酶(aconitase)、酮戊醯酸去氫酶複合體(ketoglutarate dehydrogenase complex)與琥珀酸去氫酶(succinate dehydrogenase)異常。 (4) 呼吸鏈異常(Respiratory chain defects): 已知為氧化磷酸化作用(oxidative-phosphorylation, OXPHOS)疾病或是電子傳遞鏈(electron transport chain, ETC)疾病,起因於呼吸鏈複合體I~V中,有1個或多個在基因譯碼時產生變化。呼吸鏈異常為造成先天性高乳酸血症的主因。此異常可因nDNA或mtDNA突變造成;mtDNA突變是母系遺傳,引起氧化磷酸化過程中複合體I、III、IV或V的缺失;nDNA異常為遺傳性或是偶發性,可能影響任一複合體。呼吸鏈異常時,乳酸堆積增加明顯高於丙酮酸,因此兩者比值通常超過25。如同PDC缺乏的患者,呼吸鏈異常患者腦脊髓液中的乳酸濃度高於血中乳酸濃度。 (5) 粒腺體腦症伴有乳酸血症及中風發作症(mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes, MELAS): 此症是由於粒腺體基因異所致,臨床症狀多變,患者早期發展正常,之後會有類偏頭痛,局部或全身性的抽搐,生長遲緩、聽障、四肢無力、嘔吐、血乳酸升高、運動的耐受性不足及類中風性發作的情形。
(6)肌抽躍性癲癇合併破碎紅色肌纖維症(myoclonic epilepsy and ragged red fiber disease,MERRF): 此症也是由於粒腺體基因異所致,主要症狀為漸進式運動失調、抽筋麻痺、肌無力,或是發音不良、視神經萎縮、生長遲緩、聽障及失智,孩童時期即會發病。 (7)神經組織肌肉無力伴隨運動失調與色素沉積性視網膜炎(neurogenic muscle weakness, ataxia and retinitis pigmentosa,NARP): 由於mtDNA產生變異造成,發病於兒童或青年時期,主要影響神經系統,有麻木、耳鳴、手腳疼痛,肌肉無力、運動失調、視網膜色素沉積等症狀。 (8) Leigh氏症候群(Leigh syndrome): 此症也稱為童年期腦脊髓病變,與mtDNA點突變有關,臨床症狀相當多樣化,為中樞神經退化之疾病,患者通常於兩歲前發病,臨床表現包含:視力萎縮、眼肌麻痺(ophthalmoplegia)、眼球震顫、呼吸異常、運動失調、低張力不足、抽筋、乳酸中毒與發展遲緩。 (9) Pearson症候群(Pearson syndrome): 是由於mtDNA大片段缺失造成,通常為偶發型的,患者於嬰兒期會出現生長遲緩、腸道吸收不良、胰臟功能異常、腹瀉、乳酸值升高與鐵芽球性貧血(sideroblastic anemia)。孩童患者長大後可能會以Kearns-Sayre 症候群表現。 (10) Kearns-Sayre 症候群(Kearns-Sayre syndrome): 患者通常於20歲前發病,有眼外及四肢肌肉無力、非典型色素沉積性視網膜發炎及心臟傳導障礙。血中乳酸與丙酮酸值通常會增加,即使血中乳酸為正常值,腦脊髓液中乳酸值仍高。患者若在20歲後有這些症狀,稱為慢性進行性眼外肌麻痹症(Chronic progressive external ophthalmoplegia,CPEO)。這兩項疾病的患者可能也會表現出多重內臟器官機能障礙,包括:心臟、神經系統、肝臟、肺臟及腸胃道。 (11)其他: 如粒線體肌肉病變、Barth症候群,為X-linked肌肉病變,影響心臟和骨骼的肌肉組織;遺傳性心肌病變與Alpers症也是與心肌病變有關。遺傳性Leber氏運動視神經病變(LHON),是由mtDNA缺失引起的疾病。 診斷: 由於組織缺氧或是感染等原因,出生後短時間內產生嚴重酸血症之患者,可以懷疑是因嚴重酵素缺失所導致之先天性高乳酸血症;較輕微的患者,其血中的乳酸值可能只有在壓力或特定情況下偶爾升高;腦脊髓液乳酸值比血中乳酸值更容易升高,因此在中樞神經系統內或許可當作一個較為敏感且特定的指標。利用白血球或皮膚/肌肉切片,偶爾以肝臟切片,進行酵素或分子遺傳檢驗,作進一步的診斷及分類。神經影像學檢查可協助診斷與追蹤。 治療: 目前對於先天性高乳酸血症尚無良好且有效的治療方式,多是依照患者病情給予支持療法,甚至有些症狀也無法改善;臨床上曾給生物素酵素(biotindase)缺乏患者大量生物素,或是給予丙酮酸去氫酶缺乏患者大量維生素B1治療;嚴重高乳酸血症患者在急性期可以使用重碳酸鹽或是其他鹼基來導正酸中毒情況。肉鹼、維生素B1、生物素、硫辛酸(lipoate)、維生素B2、Coenzyme Q10或維生素K、維生素C或維生素E也曾使用於治療上。若有抽筋或是心臟衰竭,則需使用抗抽筋或心臟衰竭藥物治療。
目前有一種降低乳酸的藥物dichloroacetate (DCA),認為可以治療此症,礙於更精確的藥物評估,在美國尚未問市。
此症之臨床表徵多樣化,個案間之差異性也很大,例如就發生年齡而言,可自新生嬰兒至成年期,預後因人而異,愈早發病者,預後愈不好,因此須按照患者病情而給予適當的治療與照護,以協助患者於病程中有良好生活品質及預後。 資料來源: 1.NORD Guide to rare Disorders. 462-464 2.圖片來源代謝性疾病 台灣經驗. 133-146 3.http://www.genes-at-taiwan.com.tw/genehelp (1)Mitochondrial disorder. (2)Pyruvate dehydrogenase deficiency. 4.http://ghr.nlm.nih.gov/condition=fumarasedeficiency (Funarase def.) 5..http://www.wrongdiagnosis.com/medical/merrf.htm (MERRF) 6.http://ghr.nlm.nih.gov/condition=neuropathyataxiaandretinitispigmentosa (NARP) 7.http://www.emedicine.com/ped/topic2763.htm (KSS) 台大醫院基因醫學部 李妮鍾醫師審稿 罕病基金會 醫療服務組營養師謝佳君編譯2008.12 2023年蔡孟儒/李妮鍾醫師 審閱更新 |
