| 分類代碼: 0340 |
| 疾病類別: 03 |
| 疾病名稱: 轉醛醇酶缺乏症 ( Transaldolase deficiency ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 沒有 |
|
ICD-9-CM診斷代碼:271.8 ICD-10-CM診斷代碼:E74.89 疾病簡介
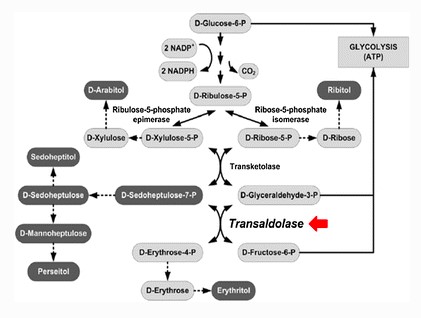
轉醛醇酶(TALDO)缺乏症(OMIM 606003)是一種罕見的磷酸戊糖途徑(pentose phosphate pathway,PPP)的先天代謝缺陷,由位於11p15染色體上的 TALDO1 基因的致病性變異所引起。磷酸戊糖途徑(PPP),存在於肺、肝、乳腺、大腦、腎上腺皮質和皮膚等組織中。 PPP分為氧化和非氧化兩個部分,負責產生NADPH和核糖用於合成核糖核酸(RNA、DNA)和三磷酸腺苷(ATP)。 TALDO是非氧化部分的第二種酶,可在PPP和糖酵解之間建立可逆連結。該酶催化三碳酮醇單元從景天庚酮糖 7-磷酸(sedoheptulose 7-phosphate)可逆的轉移到甘油醛3-磷酸(glyceraldehyde 3-phosphate),形成赤蘚糖4-磷酸(erythrose 4-phosphate)和果糖 6-磷酸(fructose 6-phosphate)。在轉醛醇酶缺乏症中,景天庚酮糖 7P 會堆積,且核糖 5P無法再循環,導致 NADPH 和穀胱甘肽(GSH)缺乏、脂質氫過氧化物(lipid hydroperoxides)濃度增加,粒線體跨膜電位喪失。因此在 TALDO 缺乏症中,許多代謝產物在尿中濃度會提高。
轉醛醇酶缺乏症其主要臨床表現通常出現在新生兒期,懷孕期間的胎兒異常發現和母體疾病並不常見,但以前也有記錄,表現為IUGR、母體HELLP症候群、羊水過少畸形、胎兒脾臟腫大和胎兒窘迫。可能表現出廣泛的臨床表型,包括胎兒水腫;肝脾腫大;肝功能障礙;血小板減少症;貧血; 腎、呼吸和心臟異常。畸形特徵包括向下傾斜的瞼裂、低位耳和皮膚鬆弛。所有發病的個案的尿液多元醇(polyol)濃度均異常,可用作診斷的生物標誌物。症狀的嚴重程度和預後因個案而異。
 發生率
此症相當罕見,估計發生率小於1/1,000,000。
病因學
缺乏轉醛酶會導致 7-磷酸景天庚酮糖增多(與正常相比,它在血液中的濃度增加了 6 到 7 倍),從而減少了5-磷酸核糖向6-磷酸葡萄糖的轉化,而這種反應對於釋放 NADPH 很重要。還原型穀胱甘肽對於調節線粒體膜通透性至關重要,並且取決於從氧化型穀胱甘肽再生的磷酸戊糖途徑產生的 NADPH。轉醛醇酶在男性生育能力中起重要作用;這是因為它維持線粒體跨膜電位及其在 NADPH 釋放中的作用。因此,轉醛酶缺乏會降低精子並降低男性生育能力。
遺傳模式
體染色體隱性遺傳。轉醛醇酶缺乏症是由 TALDO1 基因的雙等位基因突變引起的。該基因含有 17.73 kb 區域,包含八個編碼外顯子,由 337 個氨基酸組成。
臨床表徵
.產前:子宮內生長受限(IUGR)、低出生體重、羊水過少
.成長:身高體重發育緩慢
.頭頸部:三角形臉、頸部短、皮膚皺褶過多
.耳鼻喉:低耳位、凹陷鼻樑、口部寬大、嘴唇薄、突出或短咽喉
.心血管:先天性心臟缺陷、左心室擴張、卵圓孔擴大、心室、房室間隔缺損、主動脈粗大、開放性動脈導管、主動脈狹窄、先天性心臟缺陷(多數病人)
.呼吸系統:孝喘、功能性呼吸異常
.肝脾系統:肝脾腫大、小結節性肝硬化、先天性肝臟纖維化、肝臟功能紊亂、谷氨酸代謝異常
.腸胃道:產前超音波檢查可見高滲性腸道
.泌尿生殖系統:腎臟功能異常(腎小管病變、腎功能衰竭、腎鈣化)、男性外生殖器可見陰莖過小、女性陰蒂肥大
.骨頭:前囟門寬大
.皮膚:出生時額頭有皺紋(部分患者)或皮膚皺褶多
.肌肉、軟組織:新生兒水腫(在一些病人中)
.內分泌特徵:甲狀腺機能減退
.血液學:貧血、血小板減少症、全血球下降
診斷
.代謝物分析
分析尿液中尿糖和多元醇的自體酶(Autozygome analysis)可用於診斷轉醛醇酶缺乏症。可利用液相色譜串聯質譜法(liquid chromatographytandem mass spectrometry)和氣相色譜法(gas chromatography with flame ionization detection)測量尿糖和多元醇。
.基因分析
藉由分子基因診斷分析 TALDO1 基因是否有致病變異。
治療方式 目前還沒有治癒轉醛酶缺乏症的方法,可能可以考慮肝移植,但仍有疾病復發的風險。因此,現行的治療方法可能主要以使用 N-乙酰半胱氨酸(N-acetylcysteine)增加穀胱甘肽(GSH)的產生,並使用抗氧化劑(例如維生素 C 或 E)減少氧化壓力。2009 年進行的一項研究使用口服 N-乙醯半胱氨酸治療轉醛酶缺陷小鼠,可以預防與該疾病相關的症狀。
此外,該疾病的預後通常是多變的,並且患者的壽命通常由於肝功能衰竭而縮短。因此,有效的早期診斷可能會減輕病況。有文獻建議應針對患有不明原因肝病、肝脾腫大、溶血性貧血和/或胎兒水腫的新生兒、嬰兒和兒童進行轉醛醇酶缺乏症的進一步相關檢查,尤其是合併孕婦體重過度增加時。2003 年有人提出了一種篩檢方法,結果發現轉醛醇酶缺乏症患者的血片中,7-磷酸景天庚酮糖的濃度升高。 因此,該方法可與尿液中多元醇的濃度分析一起用於轉醛醇酶缺乏症篩檢。
遺傳諮詢 轉醛酶缺乏症之產前診斷仍然是一個挑戰,通常要通過基因分析來確認。然而,早期和準確的產前診斷可以帶來更好的結果,包括規律產檢和適當的產後治療。特別胎兒超音波監測,可以幫助早期識別臨床表現(如MCA-PSV升高、心臟腫大和胎盤厚度),這些都是重要的預後指標。
另外,研究指出,TALDO缺乏症是一種具多樣表現型的疾病,在調查不明原因的肝脾腫大或胎兒貧血的產前病例時,應考慮到這種疾病。雖然目前沒有具體的治療方法,但對羊水或絨毛中的TALDO1 基因進行有針對性的分子分析,仍有其幫助價值。
參考資料 1. Ada hamosh. (2015, May 7). TRANSALDOLASE DEFICIENCY; TALDOD (Hilary j. vernon & Joanna, Eds.). OMIM. https://omim.org/clinicalSynopsis/606003
2. Banne E, Meiner V, Shaag A, Katz-Brull R, Gamliel A, Korman S, Cederboim SH, Duvdevani MP, Frumkin A, Zilkha A, Kapuller V, Arbell D, Cohen E, Eventov-Friedman S. Transaldolase Deficiency: A New Case Expands the Phenotypic Spectrum. JIMD Rep. 2016;26:31-6. doi: 10.1007/8904_2015_474. Epub 2015 Aug 4. PMID: 26238251; PMCID: PMC4864716.
3. Xue J, Han J, Zhao X, Zhen L, Mei S, Hu Z and Li X (2022) Prenatal Diagnosis of Fetus With Transaldolase Deficiency Identifies Compound Heterozygous Variants: A Case Report. Front. Genet. 12:752272. doi: 10.3389/fgene.2021.752272
4. https://omim.org/clinicalSynopsis/606003
5. Al-Shamsi, A.M., Ben-Salem, S., Hertecant, J. et al. Transaldolase deficiency caused by the homozygous p.R192C mutation of the TALDO1 gene in four Emirati patients with considerable phenotypic variability. Eur J Pediatr 174, 661–668 (2015). https://doi.org/10.1007/s00431-014-2449-5
編撰:彭郁婷、郭靜維
審閱:陳薈安醫師、李妮鍾醫師
|
