| 分類代碼: 0334 |
| 疾病類別: 03 |
| 疾病名稱: 巴氏症候群 ( Barth Syndrome ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 沒有 |
|
ICD-9-CM診斷代碼:759.89 ICD-10-CM診斷代碼:E78.71 病因學: 巴氏症候群(BTHS; OMIM #302060)於1983年首次被提出。此症特徵為具有嗜中性粒細胞減少症、骨骼肌病變及心肌病變(CMP)症狀之性染色體遺傳性疾病。此症候群屬於廣義的粒線體疾病。 巴氏症候群是由於位於染色體Xq28位置上的TAZ基因突變導致tafazzin此蛋白質功能尙失所引起的。Tafazzin為位於粒線體內膜小葉的磷脂質轉酰基酶,此酶在心肌磷脂(cardiolipin)的再成型(remodeling)上扮演重要的角色。心肌磷脂(cardiolipin)對於粒線體皺摺的形成、調節組合及穩定電子傳遞鏈複合物相當的重要。它與了粒線體的細胞凋亡反應。高氧化壓力的組織如:心肌及骨骼肌通常具帶有四個亞麻酰基之心磷脂(tetralinoleoyl cardiolipin or L4‐CL)。TAZ基因突變會導致L4‐CL的形成減少、monolysocardiolipins (MLCL) 攜帶三個亞麻酰基族群心磷脂之中間產物的增加。Tafazzin會促進在主要為L4‐CL的成熟心磷脂初合成後,不成熟的心磷脂再成型(remodeling)。巴氏症候群由於tafazzin異常,導致成熟的CL較少而MLCL:L4‐CL的比例增加。 發生率: 巴氏症候群為罕見疾病,全球登錄患者少於500人,並無種族的特異性。在美國每年估計約有超過10位新的個案被診斷為巴氏症候群。雖然目前巴氏症候群實際上的流行率仍未知,巴氏症候群基金會報導指出每300,000-400,000新生兒中約有1位新生兒會被診斷出為巴氏症候群患者。最近有越來越多數據顯示巴症候群的診斷非常有可能被低估了。 遺傳模式: 為X染色體性聯隱性遺傳疾病,性聯遺傳指的是缺損的基因位於性染色體上,而隱性遺傳是指必須一對(兩條)染色體皆有缺損才表現病徵者。 女性患者身上必須有兩條缺陷的X染色體同時集中在一起,才會發病,若只被遺傳一條有缺陷的基因,則是無症狀的帶因者,女性帶原者所生育的子代中,有50%的男孩會罹患這個疾病,而有50%的女孩會為帶原者;但在男性則只需一條缺陷基因即會產生症狀。在男性患者所生育的子代中,所有的女兒則皆為帶原者。 臨床上表徵: 巴氏症候群患者會有某些或是全部被報導與疾病有關的臨床表徵。最常被報導出來的臨床表徵為心肌病變、骨骼肌病變、生長遲緩、嗜中性粒細胞減少症及尿液中3-甲基戊烯二酸(3‐ methylglutaconic acid , 3‐MGCA)排出量增加。  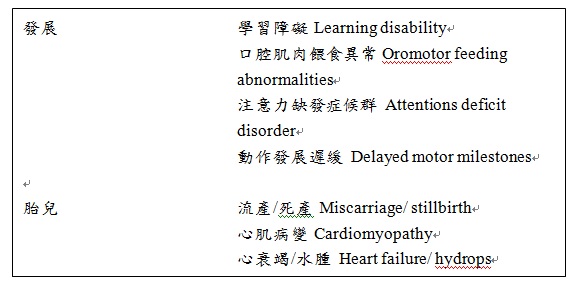 女性帶因者: 先前認為女性帶因者仍存在一條正常功能的基因,因此不會表現巴氏症候群的臨床表徵。然而,有文獻報導一名有巴氏症候群臨床表徵的女性嬰兒,在TAZ基因外顯子1-5的位置上有缺失。皮膚纖維母細胞呼吸鏈分析顯示複合體I、III及IV的活性降低。 診斷: 臨床診斷 過去,巴氏症候群主要以男性個案之嗜中性粒細胞減少症、心肌病變、骨骼肌病變及尿液中3-甲基戊烯二酸量上升等臨床表徵作為診斷的依據。然而,由於巴氏症候群臨床表徵相當的多元,若僅依賴此判斷依據往往會造成診斷上的困擾。 實驗室診斷 MLCL:L4‐CL比值 此項檢驗可在各種不同的組織上執行,包括在血片上執行。然而,雖然此檢驗於臨床上看似相當具有前景,但執行上仍有許多限制,目前並未被廣泛的使用。 目前可經由國內或國外的實驗室執行TAZ基因檢測,以幫助診斷巴氏症候群。目前大部份包含左心室致密化不全或擴張型心肌病變疾病診斷的基因檢測套組,皆有包括TAZ基因。 治療: 嗜中性粒細胞減少症 顆粒性白血球聚落刺激因子(G-CSF)已被廣泛的使用在巴氏症候群症狀的控制上。在臨床治療需要下,顆粒性白血球聚落刺激因子連同合適的預防性忼生素,為一種典型合併被用作嗜中性粒細胞減少症的控制方法。治療通常可協助增加患者體內嗜中性粒細胞數至接近正常範圍,且可藉由患者嗜中性粒細胞數的變化,判斷是否需持續使用此治療方式。根據症狀嚴重程度,患者可於每週兩次或是隔天,皮下給予顆粒性白血球聚落刺激因子。 生長遲緩 雖然在小於15歲的巴氏症候群患者有被通報其生長激素較低的情形,然而於青少年時期的後期及20歲早期,患者體內之生長激素含量會比一般人來的較高。除非已有診斷文件確立患者確實缺乏生長激素,否則並不會使用補充生長激素作為常規地治療方式。 精氨酸耗損可能牽涉導致巴氏症候群患者較慢的生長速率。目前已有相關文件指出巴氏症候群患者血清中的精氨酸含量較低,這很有可能造成患者體內蛋白質合成受到限制。增加精氨酸補充劑的使用為可能改善生長速率的一治療方式。 左心房收縮功能異常及心衰竭 與心肌功能異常有關的治療首要要務為減輕患者症狀並延長壽命。過去幾年來越來越多對於具擴張型心肌病變症狀的巴氏症候群患者之藥物及手術治療方式的改進,其中大部分的治療方式,是由其他原因用來治療成人心衰竭及兒童擴張型心肌病變而衍生出來的方法。 心律不整 雖然目前巴氏症候群患者之心肌病變受到人們很多的關注,然而心律不整這個臨床表徵可能才是造成患者死亡的主因。目前,心室性心律不整(ventricular arrhythmias)的危險性已被熟知,此表徵可能是由於代謝性酸中毒(metabolic acidosis)或伴隨左心室收縮功能不全(left ventricular systolic dysfunction)所造成。心室性心律不整造成的心室心搏過速或心室震顫,可能是導致患者心源性猝死的現象。 目前已知有巴氏症候群患者使用裝置植入性心律整流去顫器,然而其治療的有效性數據,還是非常的有限。造成生命威脅的心律不整徵狀,可能發生於任何年齡層的患者中,特別在年齡較大的患童身上較為常見。因心律不整有可能突然在任何時間點上發生,因此臨床上持續的監測如:心電圖及24小時心電圖應作為常規臨床控制的一環。 預後: 以前,巴氏症候群常被視為造成嬰兒及早期孩童死亡的疾病。由於醫療的進步,對於疾病症狀已有相當程度的改善,如嗜中性粒細胞減少症、感染風險、骨骼肌病變、心臟相關的病變的控制,巴氏症候群患者目前有較高的存活率。雖然巴氏症候群患者於4歲以前的致死率相當高,然而有些患者仍能存活到40幾歲,還其中有一位患者紀錄活到51歲。 最近有一篇有趣的報導提出出生在西元2000年之後的巴氏症患者族群,其存活率達70%,相較於出生於西元2000年之前的患者存活率20%有明顯的改善。此現象顯示改善患者症狀控制方法,確實能帶患者存活率改善的結果。 參考文獻: Jefferies JL. 2013. Barth syndrome. Am J Med Genet Part C Semin Med Genet 163C:198–205. 翻譯: 林恩如 2014.09.05 審稿: 李妮鍾醫師 2014.09.05 2015年委託台大醫院基因醫學部 審稿 以上資料轉錄自「罕見遺傳疾病一點通」網頁 |
