| 分類代碼: 1810 | |||||||||||||||||||||||||||||||||||
| 疾病類別: 18 | |||||||||||||||||||||||||||||||||||
| 疾病名稱: 遺傳性出血性血管擴張症 ( Hereditary Hemorrhagic Telangiectasia ) | |||||||||||||||||||||||||||||||||||
| 現階段政府公告之罕見疾病: 有 | |||||||||||||||||||||||||||||||||||
| 是否已發行該疾病之宣導單張: 沒有 | |||||||||||||||||||||||||||||||||||
|
ICD-9-CM診斷代碼:448.0 ICD-10-CM診斷代碼:I78.0 病因學: 是一種盛行於歐洲的體染色體顯性遺傳疾病,已定位出相關染色體缺陷位置,目前已知有4個基因與此症有關。它並非是血液凝固或凝血因子缺乏的疾病,初步已知這些基因皆與血管的生成與修補異常相關,所以會造成各種黏膜、內臟血管病變與出血的產生。 發生率: 目前估計所有種族盛行率約為1~2/10,000,此症盛行率不同地區有所差異,在荷屬安地列斯群島的發生率較高,臺灣應屬於低盛行率區域。 臨床症狀: 雖然有74%的病患可以使用電腦斷層發現肝臟動靜脈畸形,但只有約8%的病患有症狀,包含高輸出性心衰竭、肝門靜脈高壓、膽道疾病、局部結節性增生等。另外,約有18%病患會有胰臟血管病變,但通常沒有症狀。 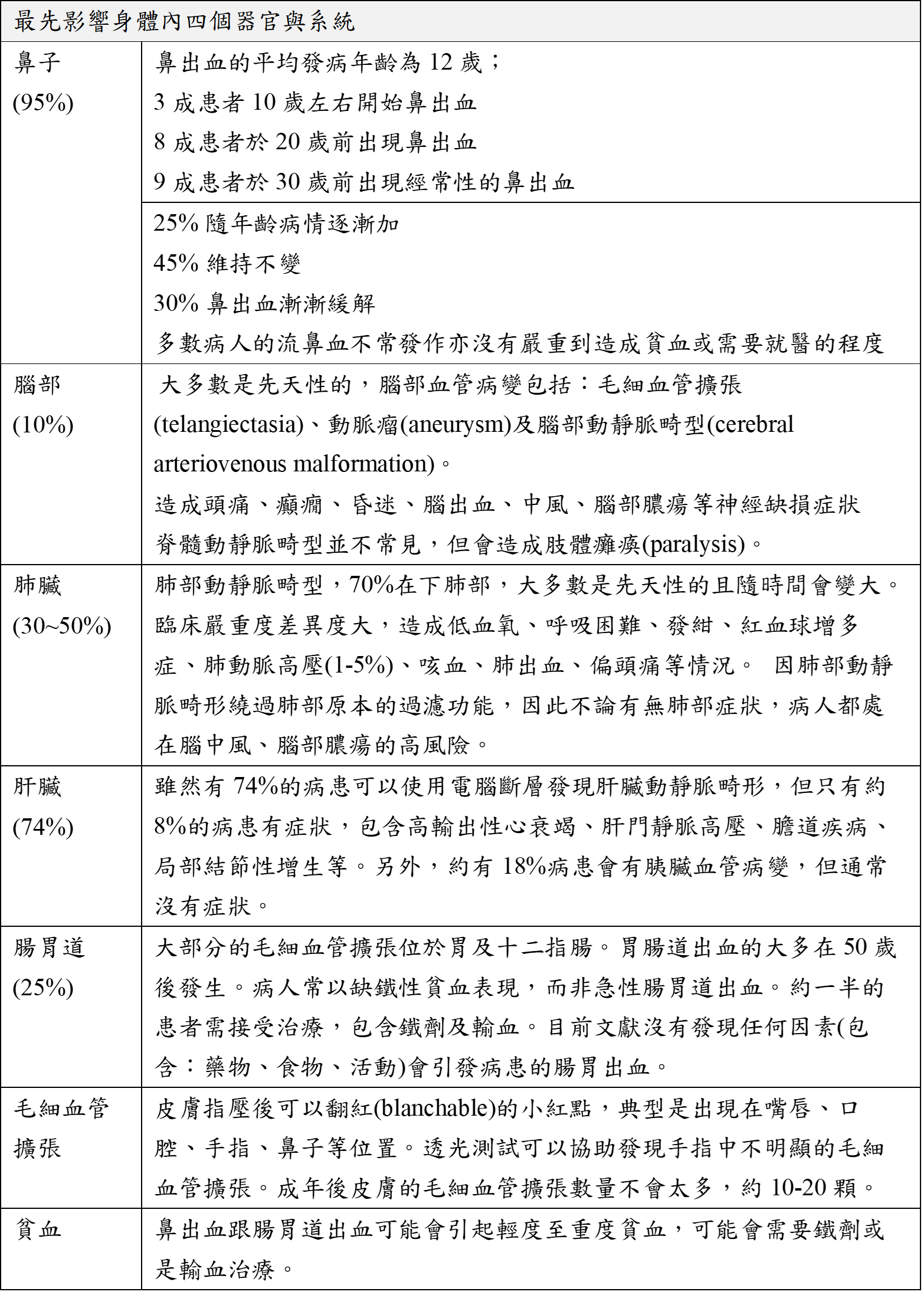 遺傳模式: 為一體染色體顯性遺傳疾病,患者的子女不分性別,皆有50%的機率會帶這缺陷的基因,若帶有這些缺陷基因則會罹病;若沒有遺傳缺陷的基因就不會罹病,相關基因表列如下,另有極少部分的患者的致病原因依然不明。
診斷: 此症的篩檢與診斷,除了目視可見毛細血管擴張外,內臟深處的血管病灶則需經電腦斷層掃瞄、核磁共振攝影術及內視鏡才能發現。 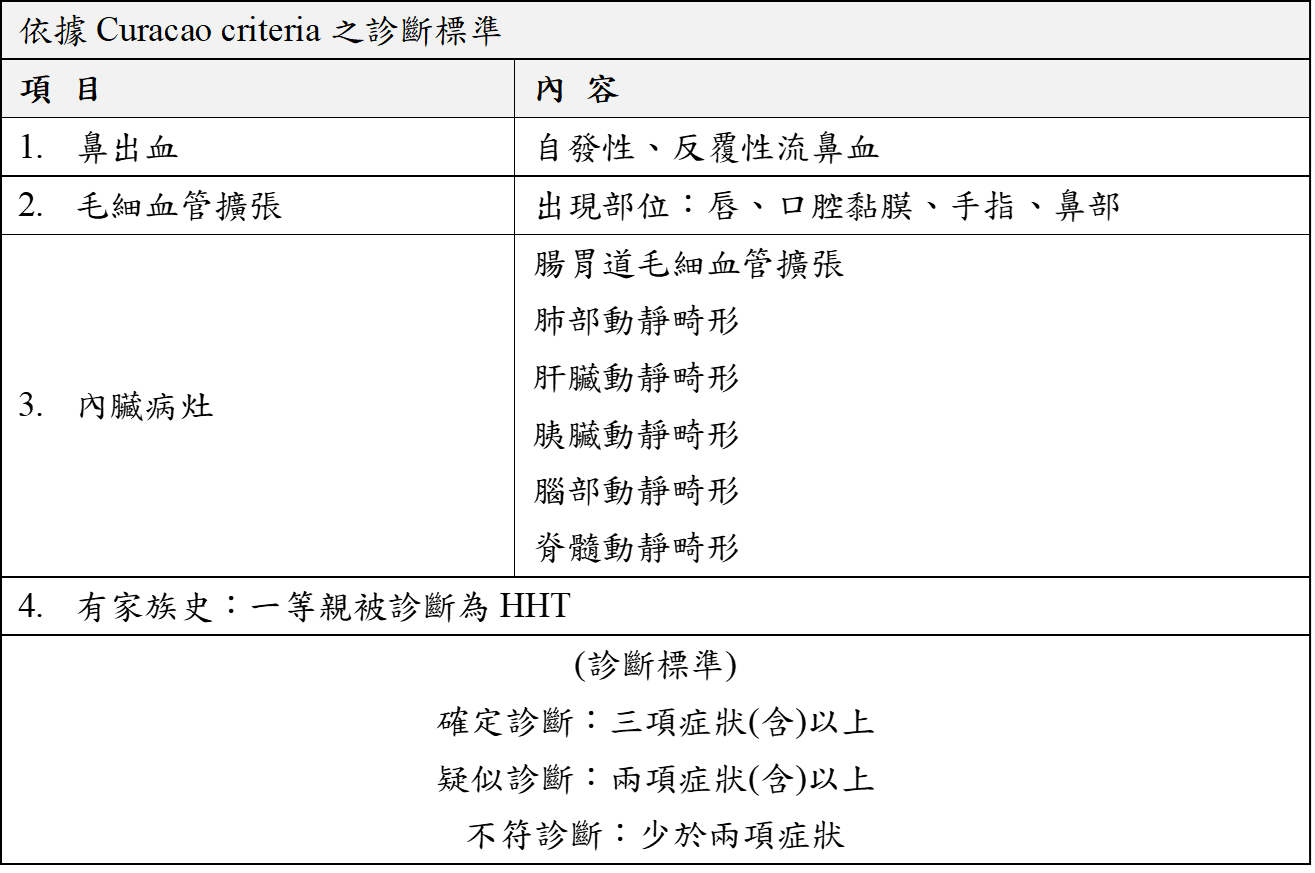 如果有發生其他症狀,應考慮其他毛細血管疾病的鑑別診斷,合併動作不協調症狀者應考慮Ataxia-telangiectasia、僅有肺部動靜脈畸形者應考慮BMPR2基因的突變、毛細血管擴張的數量多或尺寸大或非典型位置者應考慮RASA1/EPHB4基因的突變、容易出血者應考慮von Willebrand disease等凝血功能異常疾病。 治療: 因長期嚴重鼻出血而致貧血,大約10%~30%患者須長期輸血。流鼻血的處置方式,須視患者個別情況而定,簡單的可行加濕、鼻腔潤滑劑、鼻填塞止血、雷射燒灼止血,到嚴重者需行動脈結紮術。 雖然鼻中膈上皮整形手術(septodermoplasty)無法根治鼻出血,但75%的患者可以降低發作頻率與嚴重度達數年之久,若以雷射燒灼止血加上配合頰黏膜鼻中膈整形手術,緩解效果會更好。 內科的處理方式有口服或注射estrogen、progesterone等藥物,誘導鼻黏膜細胞扁平化,增加對外力的抵抗度,但對於男性患者會有性慾減退、睪丸萎縮、男性女乳症等副作用。 消化道出血可以用鐵劑治療,有必要的話可用內視鏡電燒術治療或是用手術切除出血部位及藥物治療等。 肺部的動靜脈畸形與腦部血管瘤,可行血管攝影栓塞術將病灶處血管栓塞,對於腦部血管瘤可行手術切除、血管內線圈栓塞治療,深處不易處理的血管瘤,可以採用立體定位放射手術。 由於傳統藥物治療的效果不理想或是手術風險大,因此標靶藥物的替代治療成為目前的研究方向。第二期臨床試驗指出,注射血管增生抑制藥物bevacizumab可以改善HHT病患的嚴重肝病及高輸出性心衰竭,也減少流鼻血及腸胃道出血,大幅改善生活品質。Bevacizumab第三期臨床試驗仍在進行中以更了解藥物使用的有效性及安全性。其他的血管增生抑制藥物包含tyrosine kinase inhibitors, PI3 Kinase inhibitors, Tacrolimus, Sirolimus針對HHT的臨床試驗亦在進行中。 預後: 此症應長期追蹤並定期評估貧血、血氧、內臟動靜脈畸形及息肉是否發生,一旦發現此病例,除了患者本身疾病的篩檢與治療外,家族中其餘成員也應接受長期的追蹤,以期使此症能獲得完整的醫療監測與照護。 參考資料: 1. McDonald, J., Wooderchak-Donahue, W., VanSant Webb, C., Whitehead, K., Stevenson, D. A., & Bayrak-Toydemir, P. (2015). Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Frontiers in genetics, 6, 1. 2. Robert, F., Desroches-Castan, A., Bailly, S. et al. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis 15, 4 (2020). 3. Hereditary Hemorrhagic Telangiectasia - GeneReviews 2017年委託台大醫院基因醫學部 審稿 2023年歐宗穎/李妮鍾醫師 審閱更新 以上資料轉錄自「罕見遺傳疾病一點通」網頁 |
