| 分類代碼: 1619 | |||||||||||||||||||||||||
| 疾病類別: 16 | |||||||||||||||||||||||||
| 疾病名稱: 耳-齶-指(趾)症候群 ( Oto-Palato-Digital Syndrome ) | |||||||||||||||||||||||||
| 現階段政府公告之罕見疾病: 有 | |||||||||||||||||||||||||
| 是否已發行該疾病之宣導單張: 沒有 | |||||||||||||||||||||||||
|
ICD-9-CM診斷代碼:759.89 ICD-10-CM診斷代碼:Q87.0 病因學: 泛耳-齶-指(趾)症候群(Otopalatodigital Spectrum Disorders),乃是以骨骼發育不良表徵的一群疾病統稱,包括:耳-齶-指(趾)症候群第一型(Otopalatodigital syndrome type I, OPD1)、耳-齶-指(趾)症候群第二型(Otopalatodigital syndrome type II, OPD2)、額骨骺發育異常(Frontometaphyseal dysplasia, FMD)、Melnick-Needles症候群(Melnick-Needles syndrome, MNS)及終端骨發育不良伴隨皮膚色素缺陷(Terminal osseous dysplasia with pigmentary skin defects , TODPD)。 以男性患者來說,其疾病的嚴重度可分為較輕微的OPD1,及較為嚴重的FMD及OPD2;若為MNS則常在出生前,即造成胎兒死亡。而女性患者的臨床表徵,則是有不同程度的嚴重性。 五種主要類型:OPD1, OPD2, FMD, MNS和TODPD分述如下: 1.耳-齶-指(趾)症候群第一型(OPD1)的多數患者,常是在一出生即表現症狀;此症女性患者的疾病嚴重度,可能與男性患者一樣嚴重,但少數也有只出現輕微症狀的女性患者。
2.耳-齶-指(趾)症候群第二型(OPD2)及額骨骺發育異常(FMD),女性患者疾病嚴重度就相對男性來得輕微,而多數OPD2的男性患者常是在出生一年內,即因胸廓發育不良造成肺功能不全而死亡,能夠存活超過一歲的男性患者,常會有發展遲緩的情形,而需要呼吸系統及餵食上的特別照護。
3.FMD的男性患者通常不會出現進行性的骨骼發育不良,但可能發生關節攣縮及手腳肢體的變形,而無論男性或女性患者均可能發生進行性的脊柱側彎(progressive scoliosis)。
4.Melnick-Needles症候群(MNS)來說,此症患者之間的臨床表徵差異性大,有的個案可能成人時期才被診斷出來,但其他個案可能早期便需要呼吸上的特殊照護,且壽命也比較短。MNS男性胎兒也常於在產前就死亡。
5.終端骨發育不良伴隨皮膚色素缺陷(TODPD)則是一種僅有女性會罹患的疾病,其特徵是終端骨骼的發育異常、皮膚色素缺陷及復發性指(趾)纖維瘤(digital fibromata)。
除骨骼上的異常之外,Oto-Palato-Digital Syndrome患者尚可能出現特徵性面容(包括突出的眶上脊、下斜的瞼裂、眼距過寬、寬鼻樑和寬鼻尖、顎裂、牙齒發育不全、牙齒少);腦水腫;傳導性和感音神經性聽力損失;聲門下狹窄伴隨先天性喘鳴;心臟間隔缺損、右心室流出道阻塞性病變;臍膨出; 輸尿管及尿道狹窄伴隨腎積水、尿道下裂;肌肉組織發育不全等等。絕大多數患者經家族譜評估,符合性染色體隱性遺傳的模式,是為母系遺傳,至今還沒有父傳子的案例報告出現。
目前造成此症候群的基因已知為X性染色體q28位置上的FLNA(Filamin A, alpha)基因,也是目前唯一已知的致病基因。大多數OPD1、OPD2、MNS、TODPD可以找到FLNA的致病突變;而FMD偵測率較低,約有7成左右患者可以發現突變。FLNA基因點位上的致病突變主要會造成腦室旁節結異位(periventricular nodular heterotopia),此為一神經元移行障礙疾病(neuronal migration disorder),將使患者出現癲癇、輕微的認知障礙及側腦室神經元異位。 FLNA基因轉譯後的產物為Filamin A(肌絲蛋白),是一種肌動蛋白結合蛋白(Actin-binding protein),此蛋白可藉著與integrins(為一種細胞附著因子)、跨膜接受器複合體(transmembrane receptor complexes)及次級傳遞子(second messengers)的相互作用,來調控肌動蛋白細胞骨架(Actin cytoskeleton)的重整。此症大多導因於FLNA基因之錯義突變(missense mutaion)或基因片段的微小缺損,影響肌絲的調節與功能。 泛耳-齶-指(趾)症候群是以性聯遺傳模式遺傳給下一代帶有FLNA缺陷基因的女性帶因者,其下一代若為男生,會有1/2的機率遺傳到缺陷基因而為患者;女生也會有1/2的機率遺傳到缺陷基因;而男性患者不會將致病基因傳給兒子,但女兒會遺傳到此基因而成為帶因者。 再者依疾病不同,OPD2的男性患者通常不孕,因而不會遺傳給下一代。OPD1及FMD的男性患者,則是會將致病基因傳給女兒,但並不會傳給兒子。曾生下MNS患者,並帶有FLNA缺陷基因的女性帶因者,之後每次生育都仍有50%的機率再將缺陷基因傳遞下去;若胎兒為男生,通常在出生之前或出生前後死亡;若為女生,則有不等程度的臨床表徵。因此建議高危險的家族成員,懷孕前應接受遺傳諮詢及相關的基因檢測服務。
發生率: 此症的發生相當的罕見,據估計應為十萬分之一以下。 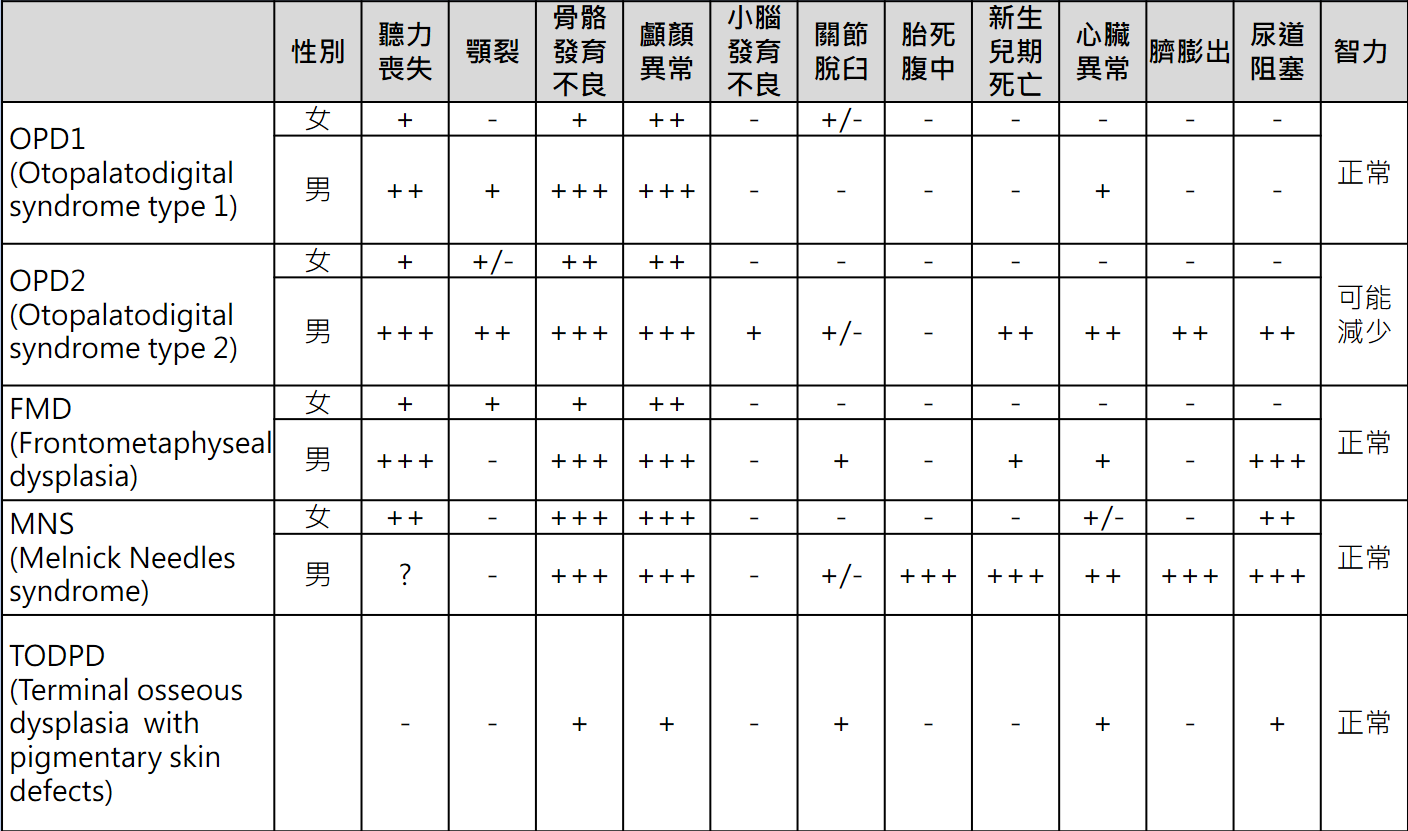 +/-:僅有少數案例 -:不常見 診斷:
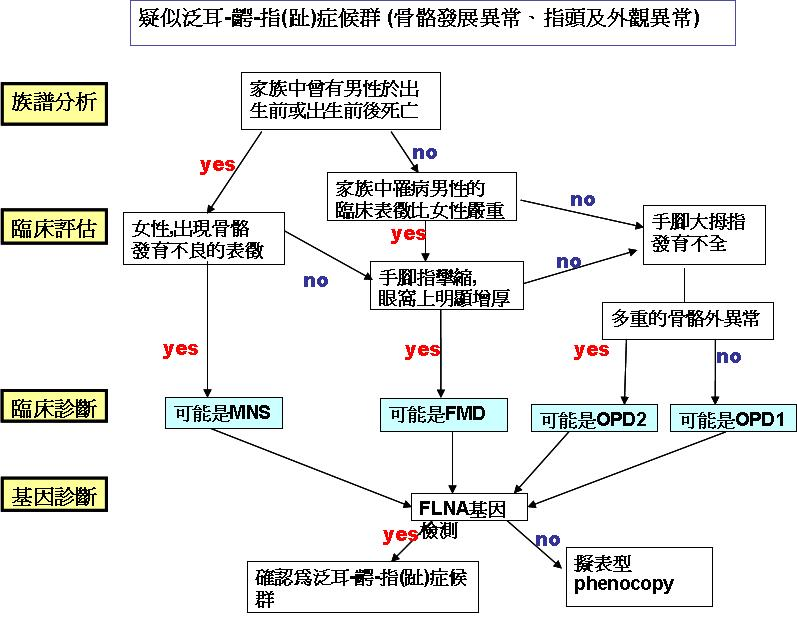
在診斷上主要為遺傳專科醫師根據個案的臨床表徵進行診治,並配合相關影像學檢查、家族譜分析,此外可經分子生物技術進行基因檢測(診斷建議流程可參考下圖)。FLNA為目前已知與此症相關的的基因,相關檢測內容如下:
 FLNA基因檢測內容
治療: 目前尚無治癒的方式,主要根據患者出現的症狀提供支持療法,助聽器可以幫助聽力喪失的患者,手足畸形、肢體彎曲、脊椎側彎嚴重、胸廓發育不全或額-眶骨畸形(fronto-orbital deformity)的個案可考慮接受手術的矯治,連續氣道正壓呼吸器(CPAP)跟下頜牽引可以改善氣管相關的併發症(如呼吸暫停、喉部狹窄等等)。
需要定期監測聽力喪失跟骨科併發症的發生,包括脊椎側彎或是顱縫早閉等症狀。
參考資料:
1. Stephen P Robertson: Otopalatodigital syndrome spectrum disorders: otopalatodigital syndrome types 1 and 2, frontometaphyseal dysplasia and Melnick-Needles syndrome. European Journal of Human Genetics (2007) 15, 3–9.
2. GeneReviewer : https://www.ncbi.nlm.nih.gov/books/NBK1393/以上資料轉錄自「罕見遺傳疾病一點通」網頁
2017年委託台大醫院基因醫學部 審稿 2024年王仲興醫師 審閱更新 以上資料轉錄自「罕見遺傳疾病一點通」網頁 |
