| 分類代碼: 1604 |
| 疾病類別: 16 |
| 疾病名稱: Cornelia de Lange氏症候群,狄蘭氏症候群 ( Cornelia de Lange Syndrome ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 有 |
|
ICD-9-CM診斷代碼:759.89 ICD-10-CM診斷代碼:Q87.19 病因學: 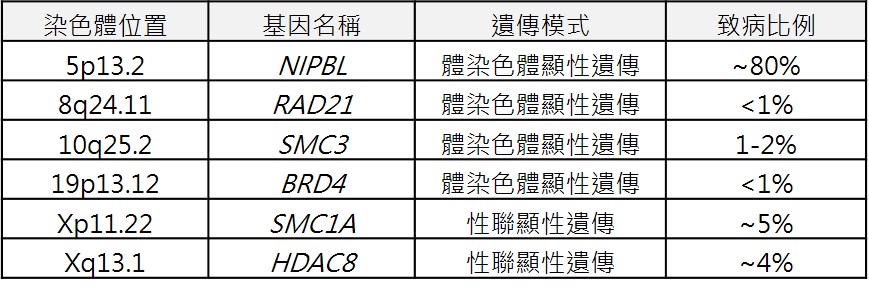 遺傳模式:
體染色體顯性或性聯顯性遺傳疾病,但大多數的個案為偶發性,並沒有明顯的家族病史,也就是由父母傳給子女的狀況並不常見。偶發的基因變異可能會遺傳給下一代,目前已有文獻報導有臨床症狀較輕型或是生殖細胞鑲嵌型 (本身無症狀,只有生殖細胞變異) 的患者生下罹病的後代。
發生率:
此症新生兒的發生率約為一萬到三萬分之一間,歐洲的統計資料則約為五萬分之一,而台灣目前並沒有確切的統計數據。
臨床症狀:
1. 特殊臉部特徵:此症最明顯的臨床表現,包含頭圍過小、一字眉、拱型眉、長睫毛、前後髮線過低、低位耳、鼻樑塌陷、短而上翻的鼻子、長人中、高拱型顎、細而下翻的嘴唇、小頷、長牙慢、齒間空隙過寬、及短頸等。
2. 生長遲緩:低出生體重及身材矮小。
3. 發展遲緩及智能障礙:嚴重的語言發展遲緩、輕度到中度的智能障礙 (平均智商為53)、有自閉、自我傷害、過動或睡眠障礙等傾向,多數患者需要進入特殊教育中學習。
4. 肢體異常:最常發生在上肢,包括前臂缺損、手指不全、小手小腳、短指(趾)、小拇指向內彎曲、第二、三腳趾併趾、或先天性橈尺骨融合。
5. 其他:多毛症、餵食困難、胃食道逆流、先天性心臟病、聽力喪失、眼睛異常 (眼瞼下垂、眼球震顫、高度近視)、及癲癇等。
診斷:
根據臨床表現來懷疑診斷,特別是擁有此症特殊臉部特徵合併生長遲滯及智能障礙的兒童,應考慮進行基因學檢查來確定診斷,如系列單基因 (serial single-gene) 、多基因套組 (multigene panel) 或全外顯子體定序 (exome sequencing) 檢測。如果與此症相關的臨床症狀和身體特徵非常輕微,則會增加診斷上的困難度。
產前診斷:
對於患有此症個案的家族,高層次超音波可以追蹤胎兒的生長狀況並評估胎兒四肢、心臟、橫膈膜、上顎、及其他器官或結構的發育情形,提供臨床參考。若是已知家族中致病性的基因變異點位,則可針對羊水中的胎兒細胞進行基因點位變異確認。
治療及預後:
目前狄蘭氏症候群並無特殊且有效的藥物治療方式,不過若無嚴重異常的狀況,其可以正常的存活,而導致患者死亡的主要原因為心臟、呼吸道、及腸胃道問題,近2/3發生於出生一年內或兩年內。患者在早期成長過程中,若有餵食困難、聽力缺損、先天性心臟病、呼吸道阻塞、及腸胃系統異常等相關問題,則需由醫療團隊定期評估監測及早期介入治療。發展遲緩與智能障礙方面,可經由早期療育的協助,如語言、職能和物理治療等,越早介入幫助越大。患者的教育程度依其潛在的智能決定,雖有患者能於一般學校就讀,但絕大多數的個案則需要特殊教育與照顧。教育策略應強調視覺教學優於傳統口語講授,如利用電腦、借助圖案或符號的擴大溝通器材,也可以透過手語的運用來克服因語言發展遲緩所帶來的挫折感。同時藉由擺動、游泳、和跳躍等可刺激前庭系統的活動,幫助患者保持愉快的心情。
相關支持團體 美國狄蘭氏症候群基金會http://www.cdlsusa.org/ 參考資料: 1. Matthew A Deardorff et. al. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. 2005 Sep 16 [updated 2020 Oct 15]. 2. National Organization for Rare Disorders (https://rarediseases.org/rare-diseases/cornelia-de-lange-syndrome/) 本會遺傳諮詢員 整理 2017年委託台大醫院基因醫學部 審稿 2022年李振豪醫師審閱更新 |
