| 分類代碼: 1005 |
| 疾病類別: 10 |
| 疾病名稱: 原發性變形性骨炎 ( Primary Paget Disease ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 沒有 |
|
ICD-9-CM診斷代碼:731.0 ICD-10-CM診斷代碼:M88.0、M88.1、M88.811、M88.812、M88.819、M88.821、M88.822、M88.829、M88.831、 M88.832、M88.839、M88.841、M88.842、M88.849、M88.851、M88.852、M88.859、 M88.861、M88.862、M88.869、M88.871、M88.872、M88.879、M88.88、M88.89、M88.9 病因學:
為一群因不同缺陷基因(heterogenous)所致的骨骼代謝異常疾病,主要因活化的蝕骨細胞(activated osteoclasts)過度進行骨骼再吸收及合成而導致骨骼病變,使骨骼變得更大、更易脆,讓患者的骨骼可能會變形、容易骨折,任何種類的骨頭都可能受到影響,特別是脊椎、骨盆、頭骨或腿部的骨骼。此症常於成年人發生,通常會在中年或更晚以後才會發病,但也有些青年發病的類型,目前已知原因如下: 成年型 一、 第二型(Paget Disease of Bone 2, PDB2):位於染色體18q21.33位置上TNFRSF11A (RANK)基因缺陷。
二、 第三型(Paget Disease of Bone 3, PDB3):位於染色體5q35.3位置上SQSTM1(Sequestosome 1)基因缺陷,最常見的。
三、 第四型(Paget Disease of Bone 4, PDB4):基因變異位於5q31位置上。
四、 第六型(Paget Disease of Bone 6, PDB6):基因變異位於1q21.3位置上ZNF687(Zinc Finger Protein 687)基因的變異。
*曾經有文獻報告第一型 (Paget Disease of Bone 1, PDB1) 與第六對染色體6p的位置有關,但後續的研究無法確認此症與6p位置突變有關聯。而目前待確認的關聯有CSF1 (位於1p13)、OPTN (位於10p13)、PML (位於15q24)、RIN3 (位於14q32)、NUP205 (位於7q33)、TM7SF4 (位於8q22)…等基因。
青少年型
因染色體8q24位置上TNFRSF11B基因(Tumor Necrosis Factor Receptor Superfamily, Member 11B)缺損,造成骨保護素(osteoprotegerin)的缺乏,而導致此症青少年型(juvenile Paget disease)的發生。
1964年Fanconi等人於一巴西裔男孩的X光及病理檢查發現,認為此症有別於osteochalasia desmalis familiaris,並之後由Stemmermann (1966)將此症稱為家族性骨異常擴張症(familial osteoectasia)。
Chong等人(2003)針對8位青少年型患者進行基因檢測,發現其中5位為同合子型的TNFRSF11B基因缺陷,研究者推論,雖非全部個案但TNFRSF11B基因缺損,仍為造成青少年型變形性骨炎的主因,且其具有可區辨的基因型-表現型(genotype-phenotype)之間的關係。 遺傳模式:
成年型
傾向為體染色體顯性遺傳模式;過去曾有不少報告論及此症在家族間遺傳的情形,甚至於綿延發生了數代,McKusick(1960)即於其報告中論述了37個家族案例。Leach等人(2001)的回顧研究亦發現,此症的發生具有家族聚集性(familial aggregation)的特徵。Morales-Piga等人(1995)在其研究中則發現,約40%患者的第一等親亦患有此症,因此經相關的家族研究發現,而認為此症傾向是以體染色體顯性遺傳模式傳給下一代。
青少年型 傾向為體染色體隱性遺傳模式;若父母為帶因者,其下一代不分性別,每一胎約有25%的機率生下此症患者。
發生率: 此病主要影響白種人,全世界以英國的發病率最高, 55歲以上大約有1–2 %的人受影響,也常見於西班牙、義大利和法國以及歐洲人移民到澳洲、加拿大、紐西蘭、南非和美國。 相反地,此病在斯堪地那維亞半島並不常見,在印度、中國、日本及其他遠東國家則很罕見。
臨床症狀:
成年型
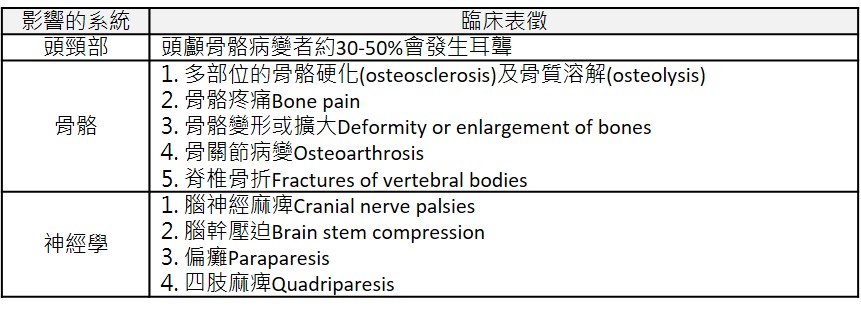
好發於40歲以上的成年人,主要為中軸骨骼(axial skeleton)發生病變,約5%的患者需要接受治療,最常見的原因為骨骼疼痛、擴大及變形。其他表徵則包括易發生骨折、耳聾及神經學上的病變。
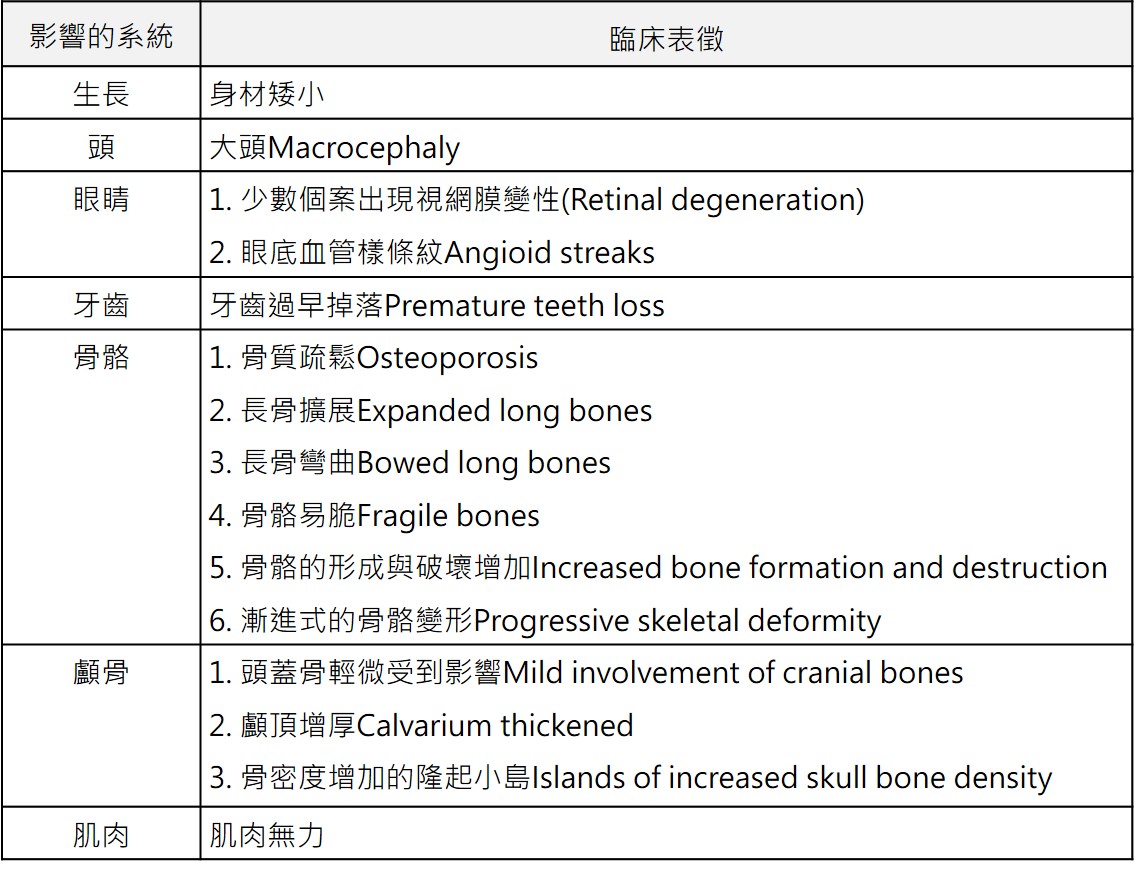
 青少年型 過去發表此型家族案例的研究中發現,此型患者在幼年時期即表現出大頭(large head)及肢體末端擴大且彎曲的表徵,再者發現到血中鹼性磷酸(alkaline
phosphatase)上升、長骨發生伴隨有骨質疏鬆(osteoporosis)及粗糙的小樑形成(coarse trabeculations)、顱頂(calvaria)顯著地增厚並具有骨密度增加的隆起小島(islands),並可能突然出現肌肉無力。 診斷:  2.攝影檢查 2017年委託台大醫院基因醫學部 審稿 2023年邱偉益醫師審閱更新 以上資料轉錄自「罕見遺傳疾病一點通」網頁 |