| 分類代碼: 0908 |
| 疾病類別: 09 |
| 疾病名稱: 肌小管病變 ( Myotubular Myopathy ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張: 有 |
|
ICD-9-CM診斷代碼:359.0 ICD-10-CM診斷代碼:G71.220 病因學 肌小管病變(Myotubular Myopathy)是先天性肌肉病變的一種,此疾病主要是肌原纖維組成肌小管時發生障礙,導致肌小管形成不良,因而使肌肉組織發展中斷並停留在胎兒時期的肌肉組織,也因此病患肌肉切片可發現肌肉細胞中,細胞核並未如同成熟的肌肉組織散佈在肌肉纖維周圍,而是位於肌肉細胞的中央,所以肌小管病變又稱為中央核肌肉病變(Centronuclear Myopathy)。
肌小管病變因致病基因不同,分成不同類型:(一)X染色體性聯遺傳型(簡稱為XLMTM),發病年齡約在新生兒時期或嬰兒時期,主要症狀為呼吸衰竭、肌肉無力及肌張力低下;(二)體染色體隱性遺傳型;(三)體染色體顯性遺傳型。第二及第三類型通常較為少見,患者肌肉會緩慢退化,發病時間從出生到成年早期都有可能出現,症狀也因人而異,即使是同一家族症狀嚴重程度也不同。
遺傳模式
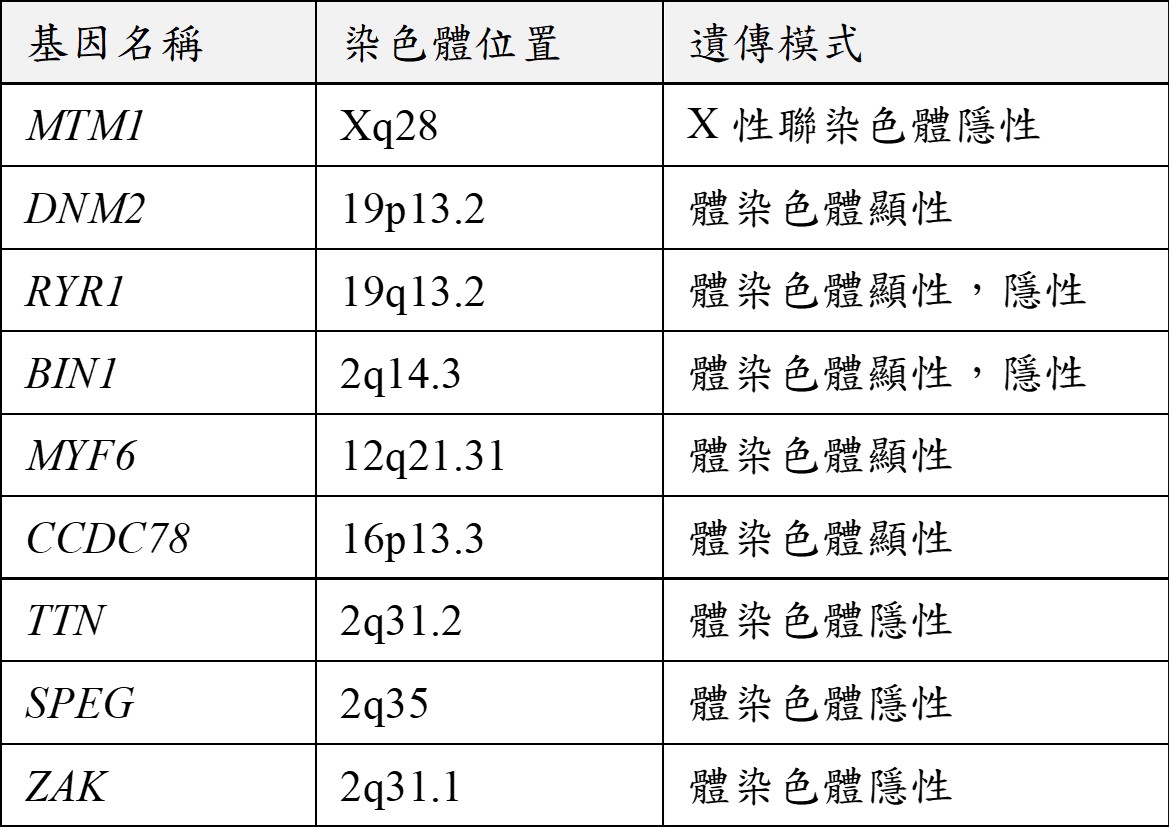
此疾病根據不同類型而有不同遺傳模式。X染色體遺傳型代表致病基因位於X染色體上,目前已知為位於Xq28的MTM1基因發生變異所致,此類型多透過女性帶因者遺傳給後代男性患者,女性帶因者並無任何症狀。其他兩型為體染色體遺傳,顯性遺傳型大多具有家族史,相關基因列於附表。
 發生率
肌小管病變很罕見,其確切的盛行率和發生率目前尚不清楚。對於X染色體性聯遺傳型(XLMTM),發生率估計為每五萬名男性新生中有1人。
臨床症狀
(一)X染色體遺傳型
此型又依症狀嚴重程度不同而細分為三型,分別是嚴重型、中間型及輕微型。最嚴重型大約一歲左右便會離開人世,但若使用呼吸治療,可改善病症並延長壽命。患童除呼吸衰竭外,智能正常,但肌肉發展遲緩及吞嚥困難,其他併發症狀包括水腦、血液方面的問題(例如球型紅血球症所引起的貧血等)、生殖器問題(如隱睪)、脊椎側彎、髖關節和膝蓋關節畸形及牙齒錯位咬合,部分患者有肝臟功能、耳部及呼吸道感染、癲癇和腎結石、膽結石等,患者臉部外觀可發現狹長的臉頰伴隨肌肉無力、眼瞼下垂、狹長的臉型及上顎高拱的特徵;另外,患者一般發育正常,身高大於平均身高九十百分位,但體重較輕。大致來說,X染色體遺傳型的肌小管病變並不屬於退化型肌肉病變,有些患童有機會緩慢地增強肌肉功能。
(二)體染色體遺傳型
此型的發病年齡從出生到成年早期都有可能,症狀也較輕微,屬於漸進式退化疾病。隨著時間的推移,肌無力的症狀會緩慢惡化,並可能導致運動發展延緩,有一些患者因為肌肉萎縮惡化而可能需要輪椅協助;不過在極少數的情況下,肌肉無力的狀況會隨著時間而改善。部分患者仍必須仰賴呼吸照護。患者面部的可能會有下垂的眼瞼和其他面部肌肉無力。其他症狀包括足部異常、高拱形顎裂和脊椎側彎。極少數的患者會發生心肌症、神經病變跟智能障礙。
診斷
利用肌肉切片的病理分析,可診斷患者是否罹患此疾病。患者的肌肉切片中可發現細胞核聚集於肌肉纖維中,而非散佈於肌肉纖維四周。另外,利用免疫染色分析法可發現肌肉纖維中,有如同胎兒肌肉的特異蛋白質(如desmin及fetal myosin等)。肌肉切片雖可診斷出此疾病,但無法達到分型的目的,若要進行分型,必須仰賴基因診斷。
治療及預後
1. 呼吸照護:此疾病的治療最重要的是呼吸照護,患者必須長期仰賴呼吸器,氣管切開術亦可使患者維持生命,但在進行手術前,建議家長們必須偕同家人審慎考慮,為患者選擇最適當的決定。在氣切之後,必須注意患童的語言訓練,可使用溝通版多加練習,以增加患童與外界接觸的能力。
2. 復健及手術治療:由於患者的呼吸問題,可配合胸腔科及復健科醫師進行呼吸訓練。另外,患童必須多加注意脊椎側灣的問題,以免影響呼吸功能,因此配合復健師的指導十分重要,若嚴重時仍需考慮以手術治療。
3. 平日預防:避免到公共場所,避免因感染而影響病情,患童若到了就學年齡,可考慮選擇在家教育。另外,定期的接受各項檢查,包括腹部超音波檢查、血液檢查、眼科及牙科的追蹤,以確實治療各種併發症狀。
一般來說,若未經呼吸治療,X染色體遺傳型的肌小管病變預後不佳。有些報導指出,此類型一歲以前的死亡率高,但在細心的照顧下,患童存活機會仍很高,家長們務必與專科醫師配合,尋求最佳的照護模式。
參考資料: 1. Soma Das, X-Linked Myotubular Myopathy: GeneReviews: https://www.ncbi.nlm.nih.gov/books/NBK1432/ 2. WallgrenPettersson C, Clarke A, Samson F, Fardeau M, Dubowitz V, Moser H,Grimm T, Barohn RJ, Barth PG. The myotubular myopathies:differential diagnosis of the X linked recessive, autosomal dominant, and autosomal recessive forms and present state of DNA studies: J Med Genet.1995 Sep:32(9):673-9 3. Claeys KG. Congenital myopathies: an update. Dev Med Child Neurol. 2020 Mar;62(3):297-302. 本會副執行長 陳冠如 編譯 國立陽明大學生命科學系 陳燕彰副教授 審稿 2017年委託台大醫院基因醫學部 審稿 2023年王麗君醫師 審閱更新 其他有關此項疾病之介紹,請詳見本會「認識罕見遺傳疾病系列」網頁 |
