| 分類代碼:0101 |
| 疾病類別:01 |
| 疾病名稱: 苯酮尿症 ( Phenylketouria, PKU ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張:有 |
|
ICD-9-CM診斷代碼:270.1 ICD-10-CM診斷代碼:E70.0 生化原理: 苯丙胺酸(phenylalanine)為人體的必須胺基酸,其主要的代謝反應為苯丙胺酸(PHE)經氧、苯丙胺酸羥化脢(PAH)、和BH4輔脢的物質作用而羥化成酪胺酸(Tyrosine)。苯丙胺酸羥化脢(PAH)存於肝臟中,是參與苯丙胺酸羥化反應的主要酵素,若此酵素功能缺乏,使得此代謝路徑上發生缺陷,將導致苯丙胺酸無法轉換成酪胺酸,而在體內大量堆積,進而產生許多有毒的代謝產物(如尿中出現大量的phenylpyruvate、phenylacetate、phenyllactate,使尿液和身體會出現腐臭味),病人若不早期治療及嚴格的控制血中苯丙胺酸之濃度於治療範圍內,將會造成腦部傷害及嚴重的智力障礙。 遺傳機制與產前診斷: 無論是典型苯酮尿症(PAH缺乏症)或四氫生喋呤(BH4)缺乏型苯酮尿症皆為體染色體隱性遺傳的胺基酸代謝異常疾病,父母雙方皆為隱性之帶原者(父母各帶一個有缺陷的基因,但沒有臨床症狀者),病患必須同時帶有兩個缺陷基因(由父母各得到一個)才會發病,故只要父母均為帶原者,則每一胎不分性別生出此症的機率為1/4。 目前可運用分子生物學(基因突變的分析)方法,對於上一胎為苯酮尿症病患及其父母,進行缺陷基因的檢測,找出並確認基因突變點後,再於母親產前或胚胎著床前進行基因篩檢,將可有效進行產前診斷,得知胎兒是否為患者。 發生率: 典型苯酮尿症(PAH缺乏症)的發生率由土耳其人的二、三千分之一,到日本的十四萬分之一,各人種及地區之發生率有很大的差異。目前台灣苯酮尿症的發生率約為四萬三千分之一。其中歐美的苯酮尿症患者約98~99%為PAH缺乏型,而國內只有約70~80%的患者為此型。國內之苯酮尿症患者約有20~30%為四氫生喋呤(BH4)缺乏型。 確認診斷: (一)建立正確的早期診斷:新生兒篩檢 在臨床上,由於出生嬰兒多無症狀,約在出生3、4個月後,堆積過高的血中苯丙胺酸代謝有毒產物才慢慢出現症狀,卻已往往造成不可逆的傷害。故如何在發病前獲得正確的診斷與適當的治療,新生兒篩檢是早期發現的最好方法。於新生兒出生48小時餵奶滿24小時後,從腳跟採取少量的血液,測定濾紙血片檢體中苯丙胺酸之含量,當濃度高於2mg/dLblood時應進一步複查,而苯丙胺酸濃度若有持續上升之現象,(高於4mg/dLblood)即應進行確認診斷。 (二)確認及鑑別診斷的項目: 1. 接受經有專科訓練之小兒科醫師的臨床評估。 2. 實驗室的確認方法為分: (1) 抽血做血液胺基酸分析,以偵測血中苯丙胺酸及酪胺酸濃度 (2) 尿液有機酸氣相層析質譜分析(GC/Mass) (3) 尿液高效液相層析定量新喋呤(Neopterin)和生物喋呤(Biopterin) ,並計算出 %B = B / N + B X 100% (4) 測定紅血球DHPR活性定量 (5) 區分PAH缺乏或BH4缺乏患者的方法包括BH4口服負荷試驗 (BH4 loading test),尿液蝶呤(urine pterin)分析,腦脊髓液神經傳導物質(CSF neurotransmitter)分析,紅血球DHPR活性定量等分析。 (6) BH4輔脢的負荷試驗(BH4 loading test):空腹給予20mg/kg,餵食一般奶粉,於0h 及24hr分別測定血中苯丙胺酸濃度及收集尿液分析Pterins的濃度,若PHE值在BH4服用後24小時下降至少30%,為對BH4有反應型,若否則為BH4沒反應型 (7) 可依血中PHE濃度高低得知病患肝臟酵素缺乏的程度,若PHE>20mg/dl則多半肝臟中酵素濃度<3%;若PHE在12-20mg/dl間,則表示肝臟仍有殘存的酵素活性。 臨床症狀: 苯丙胺酸若大量堆積在體內,在經人體代謝後產生許多苯丙胺酸相關的代謝產物,會造成病人的腦部傷害,若不早期治療將會產生嚴重的智力障礙。因為此病對腦部所造成的傷害是漸進性的,所以出生的嬰兒多無症狀,約在3-4個月後症狀才會慢慢的出現。其症狀有嘔吐、皮膚毛髮顏色變淡、濕疹、生長發育遲緩、尿液和體汗有霉臭味、抽慉、顫抖等異常的動作。若等到此時才開始治療,往往腦部神經已造成無法彌補的傷害了。 治療及癒後: (一)限制苯丙胺酸之飲食控制: 治療最好在出生兩週內開始,最遲也不可超過二個月大,以免對智力造成傷害。應限制苯丙胺酸的攝食(食物中含苯丙胺酸過多的食物:蛋、肉、魚、豆,甚至米、麵之攝取量皆需嚴格限制),最好控制血中PHE在2-6mg/dl的範圍,嚴格的飲食控制至少到6歲前(學齡前),然後可視情況放鬆控制。但建議血中PHE含量應隨時保持在10mg/dl以下,否則易影響中樞神經系統。苯丙胺酸(PHE)為人體之必須胺基酸,對典型苯酮尿症病人而言,其攝取不可太多,亦不能不足。故為補充其他人體所需的必需胺基酸,病患仍需補充一種不含苯丙胺酸之特殊奶粉,以維持身體的成長與運作,而針對此病國外已販售許多低蛋白食品,可供該類病人有更多樣的食物選擇,以改善生活品質。 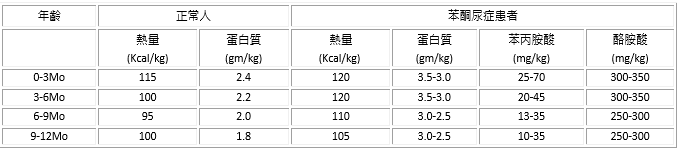 (二)治療原則 1、 針對患者給予限制苯丙胺酸之飲食控制,使血中PHE維持在2-6mg/dl的範圍,此必須有營養師的參與,除了考慮血中苯丙胺酸的進食量外,也必須考慮蛋白質等營養的均衡,使能維持正常的生長。 2、 PHE在2-8mg/dl者,則暫時不需接受飲食治療,但仍需長期密切的追蹤其血中苯丙胺酸濃度及生長發育情形。 3、 正常國人與PKU患者每日營養建議攝取量之比較 (三)建立理想血中PHE控制的目標  (四)定期監測血液中苯丙胺酸濃度,建議1歲以前每1週測1次,1至3歲每2週測1次,3歲以上穩定控制者可每1個月測1次,並於每3個月監測其營養狀況及生長發育狀況。若有發展遲緩之情形,應進行早期療育與復健。
(五)在臨床上這類病人必須終生飲食控制,早年針對許多控制到6歲後便放鬆治療控制的病人,後來證實往往易發生情緒不穩定及身心失調的症狀。而針對反抗時期的青少年,若因從眾心理而放棄飲食治療,則會造成許多行為上的偏差、暴躁等現象。尤其生育期之女性苯酮尿症患者,更應嚴格控制其血中PHE濃度,以免指數過高而生出先天性畸形兒。故控制得愈嚴格,追蹤愈密集,病患發展得狀況愈良好。 台北榮總小兒部 牛道明醫師 審稿 2015年委託台大醫院基因醫學部 審稿 2021年徐瑞聲/李妮鍾醫師 審閱更新 其他有關此項疾病之介紹,請詳見「罕見遺傳疾病一點通」網頁、本會「認識罕見遺傳疾病系列」網頁、本會「罕見疾病叢書之醫療飲食手冊」網頁、本會繪本「螢火蟲故事島第一季-娜娜的夢境」 |
|
115年公益勸募字號: 衛部救字第1141364459號 罕見之愛 溫暖同行 |
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|