| 分類代碼:0341 |
| 疾病類別:03 |
| 疾病名稱: 大腦肌酸缺乏症 ( Cerebral Creatine Deficiency ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張:沒有 |
|
ICD-9-CM診斷代碼:270.7、270.8 ICD-10-CM診斷代碼:E72.89 疾病簡介 肌酸缺乏症候群(Creatine Deficiency Syndromes ; CDS)是因先天性肌酸代謝途徑異常而致病,包括肌酸缺乏或肌酸無法運送至所需的器官。
肌酸是增加ATP的必要物質,ATP可以為人體所有細胞提供能量,故肌酸對於維持肌肉和大腦發展所需之高能量水平扮演著至關重要的角色,且也因該疾病主要是影響腦部或神經系統,故此症亦被稱為腦部肌酸缺乏症候群(Cerebral creatine deficiency syndromes; CCDS)。
肌酸缺乏症候群依不同酵素的缺乏或蛋白功能異常分成三種亞型,第一型是因肌酸轉運蛋白(SLC6A8)功能缺陷,導致肌酸轉運蛋白缺乏症(CCDS1/ Creatine transporter (CRTR) deficiency)發生,也是最常見的類型。第二、三型皆是因肌酸合成酶缺乏致病,第二型為胍乙酸甲基轉移酶缺乏症(CCDS2/Guanidinoacetate methyltransferase (GAMT) deficiency),是第二常見的類型,最後一型是左旋精胺酸甘胺酸胺基轉移酶缺乏症(CCDS3/L-arginine:glycine amidinotransferase (GATM, 又名AGAT) deficiency)。
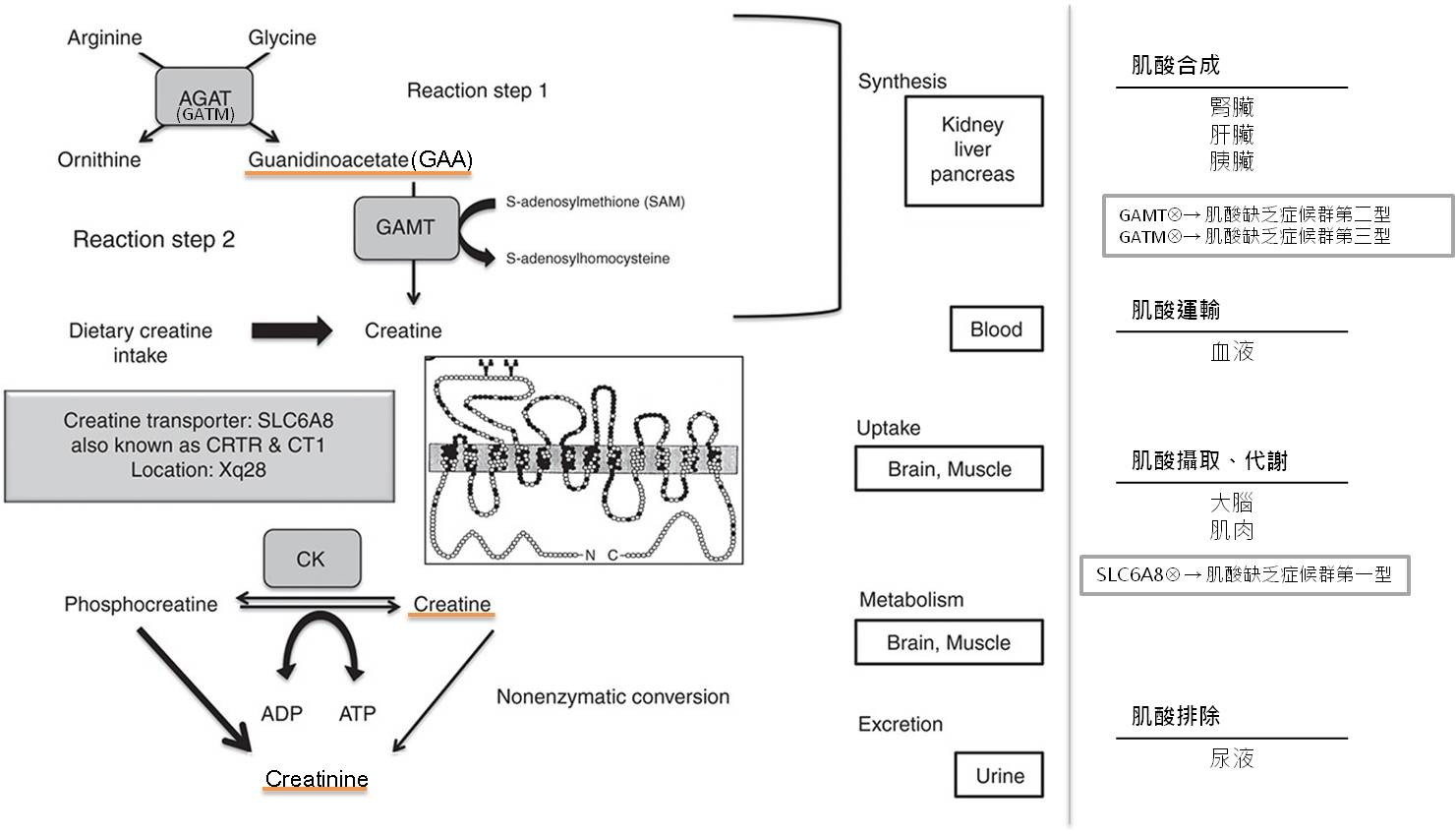 圖一、肌酸合成代謝流程[6],橘底為生化檢驗偵測之代謝物質。 常見症狀為智力障礙、語言表達遲緩、癲癇、自閉症行為,症狀之嚴重程度依類型有所區別,發病年齡也不同,CCDS1發病被診斷出的年齡介於2至66歲,代表預期壽命與一般人無特別差異,CCDS2發病年齡則介於3個月至3歲間。
病因學 肌酸缺乏症候群第一型(CCDS1)是因肌酸轉運蛋白的基因SLC6A8變異所致,該變異會使得肌酸無法被運到細胞內,導致細胞內肌酸缺乏;這會影響許多器官跟組織,特別是需要大量能量的器官,尤其是大腦。
肌酸缺乏症候群第二型(CCDS2)是因GAMT基因變異所致,GAMT基因所製造出的酵素負責肌酸合成的第二個步驟,該當基因產生致病變異會使肌酸合成異常,進而導致肌酸缺乏,還會使胍乙酸酯(具神經毒性)升高,故該類型患者症狀通常較嚴重。
肌酸缺乏症候群第三型(CCDS3)是因GATM基因變異所致,GATM基因所製造出的酵素負責肌酸合成的第一個步驟,該當基因產生致病變異會使肌酸合成異常,進而導致肌酸缺乏。肌酸缺乏症候群的基因致病變異比例分別為SLC6A8(56%)、GAMT(39%)、GATM(5%)。
遺傳模式 CCDS1為性聯遺傳疾病,是因X染色體上的SLC6A8基因產生致病變異而引起症狀;女性有兩個X染色體,若女性攜帶一個致病變異的基因,會因X染色體去活性(X-inactivation)關係導致症狀表現差異很大,從無症狀到典型症狀都有可能;而男性只有一個X染色體,若又繼承了含有致病變異的X染色體,即會罹病。
CCDS2與CCDS3為體染色體隱性遺傳。大部分父母為帶因者,各帶一個正常及一個致病的變異,通常不會出現症狀;當下一代自父母那遺傳到各自的致病變異時,就會發生體染色體隱性遺傳疾病,男女性患病風險相同皆為25%,下一代和父母一樣為帶因者的機率為50%,另有25%的機率不會帶有致病變異。
發生率 嬰幼兒期發病的肌酸缺乏症候群患者,因症狀無特異性常被誤診為腦性麻痺,兒童患者則常被診斷為自閉症或全身性發育遲緩,也導致發生率不易估計。
●目前還尚不清楚CCDS1精確的發生率,此症約佔所有無法解釋的性聯遺傳智力障礙病例的1-2%。
●CCDS2估計發生率為550,000至2,640,000中有一人患病,截至2015年,全球僅110名確診為CCDS2胍乙酸甲基轉移酶缺乏症,但在猶他州新生兒群體中CCDS2胍乙酸甲基轉移酶缺乏症的估計發生率為114,072中有一人患病。
●CCDS3目前僅有來自7個家庭共14個患者確診,因尚無患病率研究,故不清楚該類型的發生率。
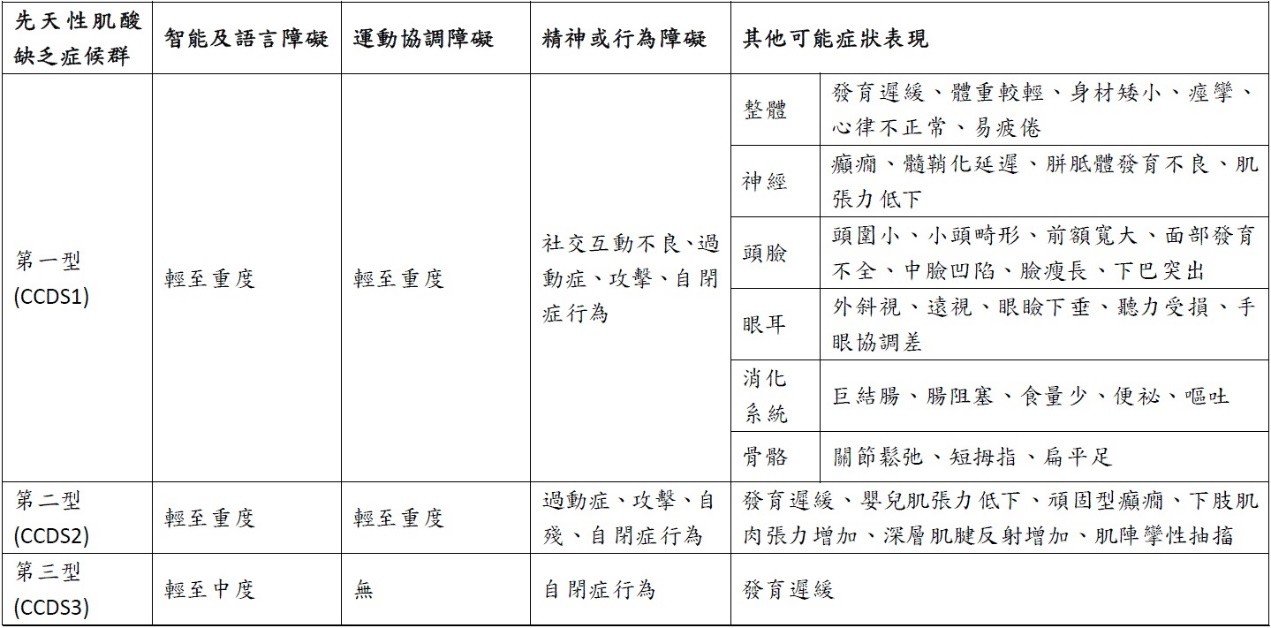
臨床表徵 肌酸缺乏症候群普遍會有整體的生長發育遲緩,呈現出智能障礙、語言發展延遲、動作發展遲緩,以及相關的神經系統疾病和行為障礙,例如注意力不足過動症(attention deficit hyperactivity disorder; ADHD),或影響溝通和社交互動的自閉症行為,另外可能偶爾有癲癇發作和腦部肌酸缺乏的症狀。
下表列出先天性肌酸缺乏症候群三種亞型可能的臨床表現,但症狀及嚴重程度皆因人而異。
 診斷方式 生化檢驗
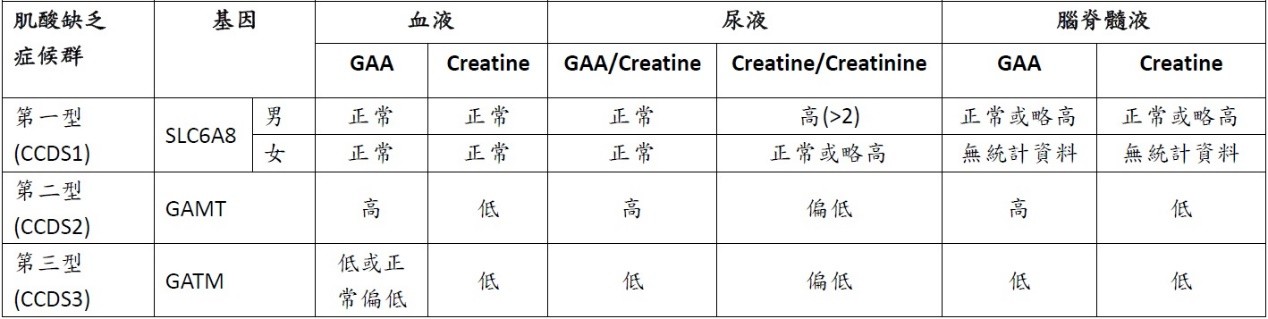
以液相層析質譜儀(LC-MS/MS)或氣相層析質譜儀(GC-MS),檢測血液、尿液、腦脊髓液或濾紙血片中的胍乙酸(Guanidinoacetate, GAA)、肌酸(Creatine)、肌酸酐(Creatinine)及相關代謝物。
(尿液是診斷CCDS1男性的最佳檢體,因為血液中肌酸濃度多為正常值,空腹採檢能有效減少飲食中肌酸的干擾。)
 影像學檢查
以腦部造影評估腦中的肌酸量,氫核磁共振光譜(H-MRS)顯示:CCDS1的男性、CCDS2、CCDS3的患者腦中幾乎沒有肌酸;CCDS1的女性帶因者腦中的肌酸量濃度則是偏低或正常。
肌酸缺乏症候群的特徵:氫核磁共振光譜(H-MRS)中有正常量的膽鹼(choline)和N-乙醯天門冬胺酸(NAA),卻完全缺乏肌酸。
分子基因檢驗
單基因定序分析:若生化檢驗結果已可初步評估先天性肌酸缺乏症候群的亞型,則可先針對三種亞型對應的SLC6A8、GAMT、GATM基因做定序分析,若是找不到會影響基因功能的變異點,可以進一步檢測涵蓋三個基因的套組分析或是全外顯子定序分析。
若已知家族致病變異,特定位點分析檢測可以鑑別出帶因者也可用於產前或胚胎植入前診檢測。
培養細胞的酵素活性檢驗
●肌酸缺乏症候群第一型(CCDS1):利用同位素標記的肌酸(Creatine)來分析轉運量(transport),從而得知纖維母細胞中CRTR的酵素活性。但可能無法準確偵測女性帶因者的酵素活性。
●肌酸缺乏症候群第二型(CCDS2):利用同位素標記的胍乙酸(GAA)來測量合成肌酸(Creatine)的量,從而得知纖維母細胞、淋巴母細胞或羊膜細胞中GAMT的酵素活性。
●肌酸缺乏症候群第三型(CCDS3):利用同位素標記的精胺酸(arginine)與甘胺酸(glycine)來測量合成的胍乙酸(GAA)量,從而得知淋巴母細胞中GATM的酵素活性。
治療 肌酸缺乏症候群第一型(CCDS1):目前尚無有效的治療法,但臨床上可補充口服肌酸(creatine)、精氨酸(Arginine)及甘胺酸(Glycine):CCDS2和CCDS3可以口服肌酸治療,以恢復大腦中的肌酸量。
肌酸缺乏症候群第二型(CCDS2) :透過低蛋白飲食限制精胺酸(arginine)和蛋白質的攝取量 。
預後及追蹤 肌酸缺乏症候群第一型(CCDS1)男性患者在補充肌酸後,並不會改善臨床結果,也不會使腦中肌酸量上升。但有研究顯示部分患者在使用高劑量的L-arginine 跟 L-glycine之後雖無法持續改善臨床症狀跟生化數值,但肌肉量與社交能力有所提升;也有一名臨床表現難治性癲癇的女性患者在大量補充L-精胺酸(L-Arginine)和L-甘胺酸(L-Glycine)後,停止癲癇發作。
肌酸缺乏症候群第二型(CCDS2)與第三型(CCDS3)的患者,若在嬰兒期的早期即接受治療是有效益的,可以預防疾病的表現。對於已有症狀的嬰兒或兒童治療,無法恢復腦部的損傷,但可以改善運動障礙,並減少癲癇發作。
對於所有患者,皆應定期追蹤及評估可能出現症狀的部位機能,如一般視力、聽力檢查,兒科評估生長發育,神經科評估運動障礙、癲癇或其他神經系統異常,骨科評估關節與運動障礙的影響等,以及是否需要配合物理治療、職能治療、語言治療或早療、特教等需求。
對於接受肌酸治療的患者,還應定期追蹤腎功能,以避免肌酸引起的腎病變。
至於確診患者其家屬若有疑慮,也建議至遺傳門診諮詢並做相關的檢查,以早期診斷並治療。
參考資料 1.Genereviews- https://www.ncbi.nlm.nih.gov/books/NBK3794/
2.OMIM-
3.NIH GARD-
4.Genetics Home Reference-
5.Sharer, J., Bodamer, O., Longo, N. et al. Laboratory diagnosis of creatine deficiency syndromes: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med 19, 256–263 (2017).
6.Clark, J., Cecil, K. Diagnostic methods and recommendations for the cerebral creatine deficiency syndromes. Pediatr Res 77, 398–405 (2015). https://doi.org/10.1038/pr.2014.203
7.NORD- https://rarediseases.org/rare-diseases/creatine-transporter-deficiency/
2024年委託台大醫院基因醫學部 審稿 |
|
115年公益勸募字號: 衛部救字第1141364459號 罕見之愛 溫暖同行 |
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|