| 分類代碼:1631 |
| 疾病類別:16 |
| 疾病名稱: 森森布倫納症候群 ( Sensenbrenner Syndrome ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張:沒有 |
|
ICD-9-CM診斷代碼:759.89 ICD-10-CM診斷代碼:Q87.5 森森布倫納症候群最初是在一對患有多頭畸形、根狀骨縮短、短指和外胚層缺陷的手足身上被報導。隨後,Levin 等人(1977)描述了來自另外兩個家族的個案,並將該疾病重新命名為顱外胚層發育不良。
森森布倫納症候群肇因於顱外胚層發育不良造成纖毛病變。臨床症狀常包含骨骼的症狀、外胚層發育異常、關節鬆弛、生長遲緩、肝纖維化、視網膜失養及具備特殊的臉部特徵。大多數罹病的兒童會產生腎臟的病變,並在嬰兒到兒童時期進展至末期腎病變。而這也是造成這些患者死亡的主要原因。在這類的患者身上也可能見到長顱的症狀(此多肇因於矢狀顱縫過早關閉)。這個主要表現為區分森森布倫納症候群與其他大多數纖毛病的依據。 病因學
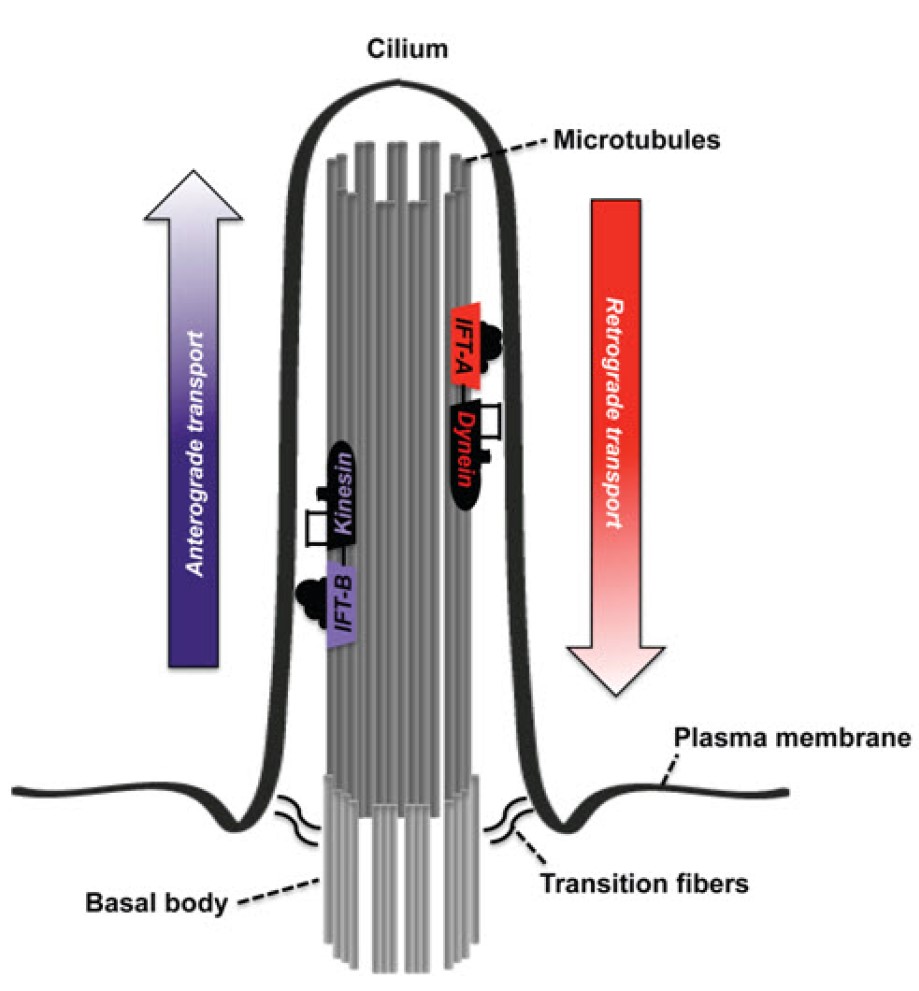
顱外胚層發育不良(cranioectodermal dysplasia)是一群纖毛病變(ciliopathies)所引起的疾病。纖毛是一種遍佈全身的細胞胞器,由微管構造(microtubule)組成,向細胞外突出,被認為有感受及傳遞胞外訊息的功能。纖毛傳遞(ciliary transport)指的是上述影響纖毛生長、訊息調控的過程,又被稱為鞭毛內運輸(intraflagellar transport, IFT)。物質及訊號的向上運輸(upward transport)須經由多重次單位的IFT-B複合體及異源三聚體的kinesin-2 motor共同作用,而向下運輸(downward transport)則由IFT-A複合體及dynein-2 motor共同作用。(如下圖)
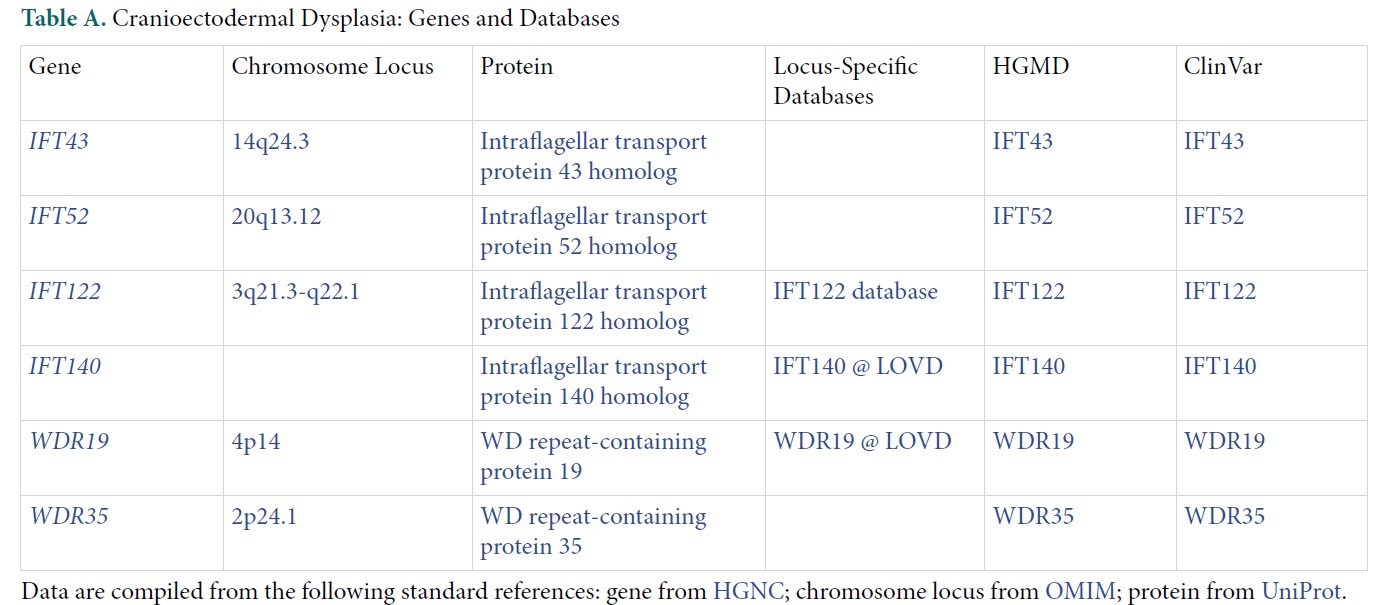
目前發現與顱外胚層發育不良相關的基因大部分都與IFT-A六聚體蛋白質複合體有關,如IFT122 (過去稱為WDR10)、WDR35(IFT121)、WDR19 (IFT144)、IFT43 (前名C14orf179)、IFT140。其他IFT-A複合體的蛋白質則由TTC21B基因所編碼。此外,負責編碼IFT139、IFT140、DYNC2H1(dynein-2 motor的次單位)等蛋白質的基因若發生突變,則可能出現與顱外胚層發育不良類似的症狀表現。
當IFT-A複合體的功能受損,纖毛的頂端會變得腫脹,且有過多的IFT-B複合體堆積。而若編碼IFT-B的基因出現突變(如IFT52),也會出現顱外胚層發育不良的表現。
 發生率
<1 / 1,000,000 (目前被報導過的個案小於一百個)
遺傳模式
體染色體隱性(autosomal recessive),相關基因如下表:
 症狀
森森布倫納症候群影響的範圍包含骨骼、外胚層(牙齒、頭髮和指甲)、視網膜、腎臟、肝臟、肺及大腦。由於森森布倫納症候群的患者仍屬罕見,對於本疾病表現型的瞭解還十分有限。以下會根據各影響的部位、器官分別作出描述:
• 典型的臉部特徵
從出生就可以觀察到,且幾乎在所有患者身上都很明顯。包括額骨突出、雙顳部變窄和前額高、低位或向後旋轉的耳朵、內眥間距過寬、內眥贅皮、下/上瞼裂、飽滿的臉頰、小頜、下唇外翻、和鼻孔前傾。
長頭畸形(頭部的前後徑較寬度增加),因矢狀縫過早閉合,會導致額葉前凸。通常於出生時即出現症狀。
• 骨骼特徵 1. 四肢
產前超音波可在懷孕中期見到多指畸形。於新生兒身上可見到中間及遠端的指骨較短且形狀異常、尺側多指 (postaxial polydactyly)、手指或腳趾間有皮膚相連。在X光下骨骺的外觀可能正常也可能呈扁平或錐狀。其他可能的發現還包括第五手指或腳趾較為彎曲、掌紋異常、手指彎曲受限、骨質疏鬆以及趾間間隔明顯等。 2. 胸腔
胸廓較窄、肋骨較短且發育不良均是常見的特徵。雖有部分患者可於懷孕中期經產前超音波診斷,但大多數仍於出生後才被發現。肋骨的異常有可能會在兒童時期恢復正常。
3. 近端長骨較短
可能於懷孕23週時發現。通常上肢又短於下肢,尤其是肱骨特別會受到影響。此外,長骨也可能呈現弓形、骨骺較扁平、幹骺端外張等狀況。
4. 生長受限
於產前或嬰幼兒時期均可能發生發育遲緩。
• 外胚層缺陷 1. 牙齒 患者乳牙通常較小、間距較寬,且出現缺牙、琺瑯質發育不全、牙齒呈現牛狀齒、融合或錐狀的牙齒。在恆齒上也會有類似的症狀。乳牙發育不良可能會影響到恆齒的發育。
2. 皮膚
妊娠中期超音波可發現蹼狀頸及頸部皮膚變厚。全身的皮膚可能較為鬆弛且出現皺摺(特別是在頸部、腳踝和手腕處),皮膚較乾且過度角質化。
3. 毛髮
嬰幼兒時期的毛髮可能較細且稀疏、髮色較淡。但也有部分病人的毛髮發育正常。
從嬰兒時期指甲就可能較正常人為寬、短,且生長較緩慢。
4. 指甲
從嬰兒時期指甲就可能較正常人為寬、短,且生長較緩慢。
• 腎臟 腎臟會出現腎小管間質性腎炎。約有百分之六十到七十的病人會出現腎功能不全。部分胎兒於懷孕中期腎臟可能會出現小的囊狀構造,可能會有羊水過多或羊水過少的狀況。出生時由於腎臟濃縮尿液的能力變差,故會出現多尿、夜尿的狀況。患者也可能出現高血壓、蛋白尿、血尿、及電解質不平衡的狀況。大多數的患者會在兩歲到六歲間進展為末期腎病變。嬰幼兒期的腎臟超音波可見到大小正常但高回音性且分化不良的腎臟。腎臟切片可見間質纖維化且有部分發炎細胞浸潤、腎小管萎縮、腎絲球硬化。部分較嚴重的個案可見到腎臟囊腫。
• 肝臟 新生兒時期的肝臟異常具多樣性。從肝脾腫大到高膽紅素血症、膽汁淤積都可能出現。在嬰兒時期可能進展至肝硬化、嚴重的膽汁淤積合併膽管增生或膽管炎。在三到四歲的幼兒可發現肝臟囊腫。若進展至肝硬化或肝臟纖維化則預後不佳。
• 眼睛 患者可能出現視網膜退化或眼球震顫。在一歲前可能出現夜盲的症狀。視網膜電圖檢測異常且眼底鏡可發現動脈變細、眼底有骨針狀沉積物。和其他纖毛病變相似,患者到二十幾歲會進展至全盲。其他可能出現的眼科問題還包括遠視、近視、內斜視、近視/遠視散光和水平眼瞼長度過長等。
• 肺部 在嬰幼兒時期因肺部發育不良或肺部感染可能出現呼吸窘迫,甚至危及生命。有些患者會出現氣喘或氣胸。隨年齡增長,肺部感染的狀況可能改善。
• 心臟構造異常 患者可能出現動脈導管未關閉、心房或心室中隔缺損、二尖瓣或三尖瓣可能較厚、心室可能較為肥厚或擴張、周邊肺動脈狹窄。
• 中樞神經系統 大多數罹病的兒童發育都正常,但仍有患者被報導有輕度發育遲緩。患者可能會在九個月到十五個月才學會坐,到三歲才學會走路。語言發展也可能較遲緩,可能在十九個月才會說少數的單詞、甚至到五歲都還不會說話。認知和社交能力通常都沒受到影響。影像學上腦部的影像可見到白質較為萎縮、腦室擴大、大腦池擴大、胼胝體發育不良、後腦室擴大、或出現丹迪沃克症候群。
• 其他 可能會出現關節較為鬆弛、或雙側腹股溝疝氣。
• 壽命 森森布倫納症候群的發病率很高,患者住院治療的機率可能上升但對於死亡率的影響尚不清楚。死亡原因可能為呼吸衰竭、心臟衰竭、低血容性休克。
診斷
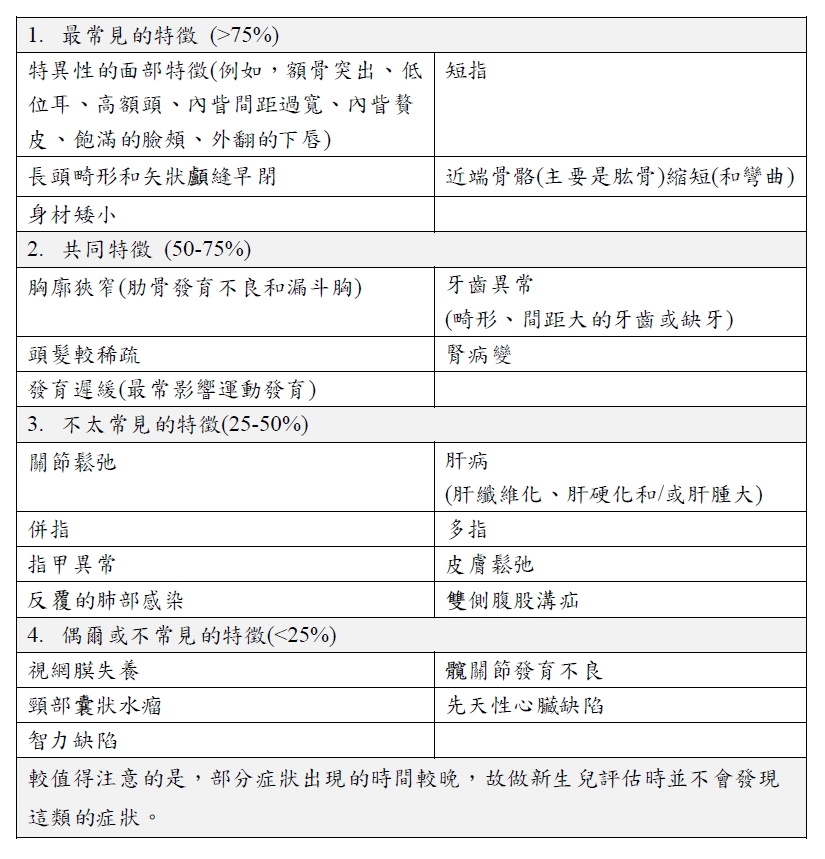
當有以下的臨床表現及影像學發現時即要懷疑其是否為森森布倫納症候群患者。其中,這些臨床表現又可因為在患者身上的常見程度分為四類(Frequent features、Common features、Less common features、Occasional to infrequent features),森森布倫納症候群的診斷可仰賴臨床診斷及分子診斷。若患者具備兩個最常見的特徵加上兩個次常見的臨床表現(至少一個的外胚層缺陷,如牙齒、頭髮、指甲的缺陷)即可做出臨床診斷。若要做出分子診斷,則可以檢驗患者是否帶有IFT43、IFT52、IFT122、IFT140、WDR19、WDR35的致病基因變異。
 治療方法
目前尚無基因治療,僅能矯正症狀及表現。如顱骨早閉、多指、心臟結構異常、腹股溝疝氣、齒列異常等,需要藉由外科手術矯正。身材矮小可補充生長激素刺激身高發展。腎臟功能異常需由腎臟科醫師長期追蹤,甚至需要腎臟移植。肝臟纖維化亦須由醫師長期追蹤,並考慮肝臟移植。肺部發育不全常需要依賴呼吸器維持,若反覆感染肺炎則需要長期服用預防性抗生素。發展遲緩的患童需要物理治療、語言治療的早期介入,以提升整體表現。目前尚無基因治療,僅能矯正症狀及表現。如顱骨早閉、多指、心臟結構異常、腹股溝疝氣、齒列異常等,需要藉由外科手術矯正。身材矮小可補充生長激素刺激身高發展。腎臟功能異常需由腎臟科醫師長期追蹤,甚至需要腎臟移植。肝臟纖維化亦須由醫師長期追蹤,並考慮肝臟移植。肺部發育不全常需要依賴呼吸器維持,若反覆感染肺炎則需要長期服用預防性抗生素。發展遲緩的患童需要物理治療、語言治療的早期介入,以提升整體表現。
參考資料
GeneReviews:
Tan W, Lin A, Keppler-Noreuil K. Cranioectodermal Dysplasia. 2013 Sep 12 [Updated 2022 Dec 15]. In: Adam MP, Everman DB, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154653/
|
|
115年公益勸募字號: 衛部救字第1141364459號 罕見之愛 溫暖同行 |
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|