| 分類代碼:0204 |
| 疾病類別:02 |
| 疾病名稱: 其他未分類之先天性尿素循環代謝障礙 ( Other Congenital Urea Cycle Disorders ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張:沒有 |
|
ICD-9-CM診斷代碼:270.6 ICD-10-CM診斷代碼:E72.20 病因學及常見臨床症狀: 尿素循環主要在肝臟中表現,為一連串移除血氨的路徑。其反應有一部份是在粒線體,有一部份是在細胞質裡面,目的是把有毒的物質(血氨),轉換成較無毒的物質(尿素),然後由尿液中排出。這個代謝路徑的任一酵素缺陷,都會導致血氨升高。另外也有許多的先天代謝性疾病,會次發性地抑制尿素循環而導致高氨血症,例如:丙酸血症、甲基丙二酸血症、異戊酸血症等。尿素循環是哺乳動物移除氮的最終路徑。在人體中,血氨主要是由麩醯胺酸(Glutamine)以及麩胺酸(Glutamate)攜帶並運送至肝臟代謝,然後藉著肝臟粒線體中的carbamyl phosphate synthetase I (CPS1)酵素反應轉化成carbamyl phosphate進而進到循環。而該酵素需要一個活性因子(N-acetylglutamate)才能完全作用,此活性因子需透過 N-acetylglutamate synthetase(NAGS)酵素的反應而產生。進到循環後,Carbamyl phosphate藉著鳥胺酸氨甲基移轉酶(ornithine transcarbamylase, OTC) 酵素作用而產生瓜胺酸(citrulline)。
瓜胺酸(citrulline)進入細胞質中再進一步藉著argininosuccinate synthetase-1 (ASS1)酵素的作用,和天門冬胺酸(aspartate)結合,產生精胺丁二酸(argininosucinate);然後被argininosuccinate lyase (ASL)酵素水解成精胺酸(arginine)和fumarate;最後,精胺酸(arginine)會被精胺酸脢(arginase, ARG1)分解,釋放出尿素(urea),而再形成鳥胺酸(ornithine)(相當於鳥胺酸為一攜帶者的角色,它不會被形成也不會消失)。尿素接著由尿液排除身體,完成正常身體代謝氮的路徑。 另外,粒線體的citrin蛋白負責將天門冬胺酸(aspartate)轉運送入細胞質作為瓜胺酸(citrulline)代謝的原料。若循環中的任何一個酵素或轉運蛋白發生問題,都可能使血氨的代謝無法順利進行而致病。 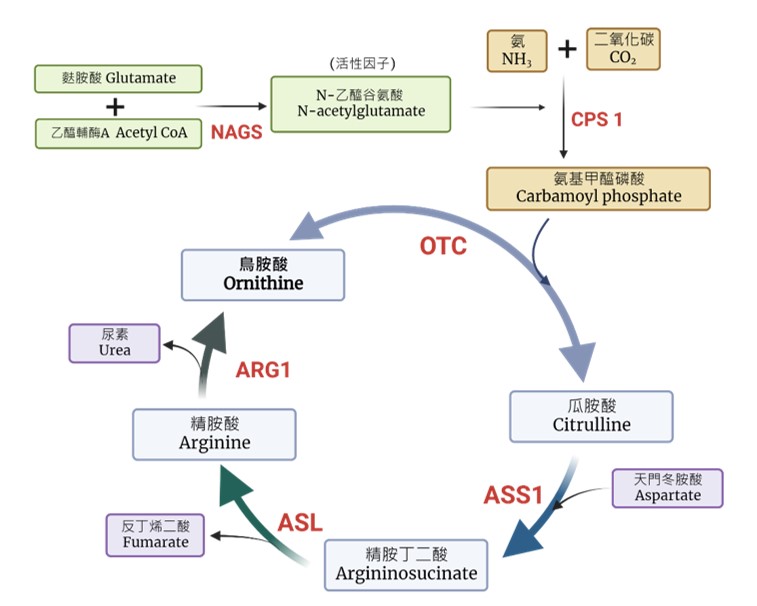 尿素循環障礙疾病幾乎可以在任何年紀表現,臨床表現嚴重度依照何種酵素缺乏及其缺乏的程度而有不同。尿素循環的上游酵素(CPS1, OTC, ASS1, ASL及NAGS等)的缺乏容易導致代謝路徑完全阻斷,病患容易在出生幾天內的新生兒時期即發病及合併高血氨症危象,例如困難餵食、活力低下、呼吸急促或緩慢、低體溫、癲癇發作、昏迷及腦水腫等,須立即住院治療。 其他酵素缺乏的尿素循環障礙疾病,在某些特定的時間患者比較可能會因為代謝上的壓力(生病、手術、長時間空腹、懷孕、暴飲暴食、精神壓力)而導致代償失調,才發生高血氨症。高血氨症的急性症狀如上述所列,但長期陣發的高血氨症可能是導致慢性神經障礙的原因,如:發展遲緩、學習障礙、行為異常及錐體症狀等。尿素循環障礙疾病的症狀大多雷同,唯有ASL缺乏症會有特殊的結節性脆髮症(trichorrhexis nodosa),argininase缺乏症會有進展性的下肢肌肉痙攣。因為早期的症狀不見專一性,診斷容易被忽略。因此測量血氨是不可少的檢查。 發生率: 各種酵素缺乏症的發生率不同,NAGS缺乏症及CPS1缺乏症的發生率極低,小於百萬分之一。ASS1及citrin缺乏症在台灣較常見的,發生率約1/10000~20000。整體尿素循環障礙疾病的發生率約為1/35000。晚發型疾病目前還沒有確切的統計資料。 遺傳模式:
大部分的尿素循環障礙疾病為體染色體(autosomal recessive)隱性遺傳疾病,表示父母親應各有此一缺陷基因的帶因,其下一代不分性別,每一胎有1/4機率罹患此症。唯有OTC缺乏症為性染色體(X-linked)遺傳疾病,對男性的影響會較大(因為男性只有一個X染色體,女性則有二個X染色體)。家族裡如果有一名成員罹病,建議尋求遺傳科醫師諮詢,以便了解下一代成員的罹病風險。
診斷:
當病人有相關的臨床症狀及高血氨症(血氨大於150μmol/L)時,須懷疑尿素循環障礙疾病;如果合併代謝性酸血症且抽血anion gap數值大於20,或有低血糖,則需要先考慮其他代謝性疾病。
反之,可以安排串聯質譜儀(Tandem Mass Spectrometry)檢測血片的胺基酸是否有尿素代謝產物的異常升高或缺少 (如: citrulline, arginine, ornithine)和其他含氮廢物的增加(glutamine, alanine, asparagine)。尿液胺基酸(homocitrulline)和尿液有機酸(orotic acid)的檢測異常亦可以提供診斷的鑑別分析。
在疾病確診上,可經皮膚切片,檢測細胞中的酵素活性,或抽血經分子生物技術進行該缺陷基因的檢測。目前也可透過抽血經次世代基因定序檢測相關的尿素循環代謝異常疾病。
目前台灣新生兒篩檢針對尿素循環障礙疾病的主要篩檢項目為citrulline, 可以提早診斷發生率較高的ASS1及citrin缺乏症。這類疾病的提早治療,輔以藥物、飲食、復健等,大多數患者的狀況都維持的不錯。
參考資料
2.Summar ML, Koelker S, Freedenberg D, Le Mons C, Ha J, Lee HS, Kirmse B., European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD). The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179–80
2022年歐宗穎/李妮鍾 醫師審閱更新 以上資料轉錄自「罕見遺傳疾病一點通」網頁、本會關於「高血氨症狀」網頁、「罕見疾病叢書之醫療飲食手冊」網頁(1)、(2) |
|
115年公益勸募字號: 衛部救字第1141364459號 罕見之愛 溫暖同行 |
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|