ICD-9-CM診斷代碼:330.0
ICD-10-CM診斷代碼:E79.8
病因學:
Aicardi-Goutieres症候群(AGS)通常最典型表現為早發性腦病,但有時會合併嚴重的智力缺損和身體障礙。有部分AGS患嬰在出生時就發現神經系統異常、肝脾腫大、肝酶素升高、血小板減少,從症狀表現來看會疑似是先天性病毒感染,但實際上並無感染發生。除此之外,大部分患病的嬰兒在出生後會有一段看似正常的發展,在之後數周不等的時間內,開始有症狀出現。個案最典型的情況是會表現出嚴重腦病的特徵,像是極度的急躁、間歇性的發燒(無感染)、癲癇、喪失生活技能和小頭症,在這個階段中,可以從患者的腦脊隨液中檢測到白血球跟與發炎相關的分子,這與中樞神經系統發炎及組織損傷表現一致。隨著病情的進展,多達40%的個案在手指、腳趾、耳朵的皮膚出現凍瘡損傷。
AGS個案近年來出現越來越多非典型的症狀,有些是較輕微的症狀,這些表現型與AGS相關基因的突變程度尚不清楚。例如,ADAR基因突變最近已經證實其臨床症狀與急性雙側紋狀體壞死有關。
發生率:
此症相當罕見,目前AGS的實際發生率仍未知。
遺傳模式:
Aicardi-Goutieres症候群為體染色體隱性遺傳為主,但也有少數屬體染色體顯性遺傳。分子遺傳學檢測應先檢測家族中的proband(發病者),找出其致病基因。AGS相關基因包括RNASEH2A,RNASEH2B,RNASEH2C,SAMHD1、TREX1及ADAR基因,屬體染色體隱性遺傳。另外,特定TREX1及ADAR基因致病點位及IFIH1基因突變,屬於體染色體顯性遺傳。
體染色體隱性遺傳模式代表每位體染色體隱性遺傳的AGS患者,其兄弟姊妹有25%的機率也會得病,50%的機會成為一名無症狀帶基因者,25%的機率不會得病,也不會成為帶基因者;若是體染色體顯性遺傳模式則代表每位顯性遺傳的AGS患者,其子女有50%的機率成為患者,另有50%的機率無症狀的帶因者。AGS患者通常沒有生育能力 。如果致病的等位基因變異在家族中已經確定,親屬的帶基因檢測及為懷孕者的產前檢查可能是必要的。
| 分型 |
遺傳模式 |
遺傳模式 |
基因 |
| AGS1 |
體染色體顯性或隱性遺傳 |
3p21.31 |
TREX1 |
| AGS2 |
體染色體隱性遺傳 |
13q14.3 |
RNASEH2B |
| AGS3 |
體染色體隱性遺傳 |
11q13.1 |
RNASEH2C |
| AGS4 |
體染色體隱性遺傳 |
19p13.13 |
RNASEH2A |
| AGS5 |
體染色體隱性遺傳 |
20q11.23 |
SAMHD1 |
| AGS6 |
體染色體顯性或隱性遺傳
|
1q21.3 |
ADAR |
| AGS7 |
體染色體顯性遺傳 |
2q24.2 |
IFIH1 |
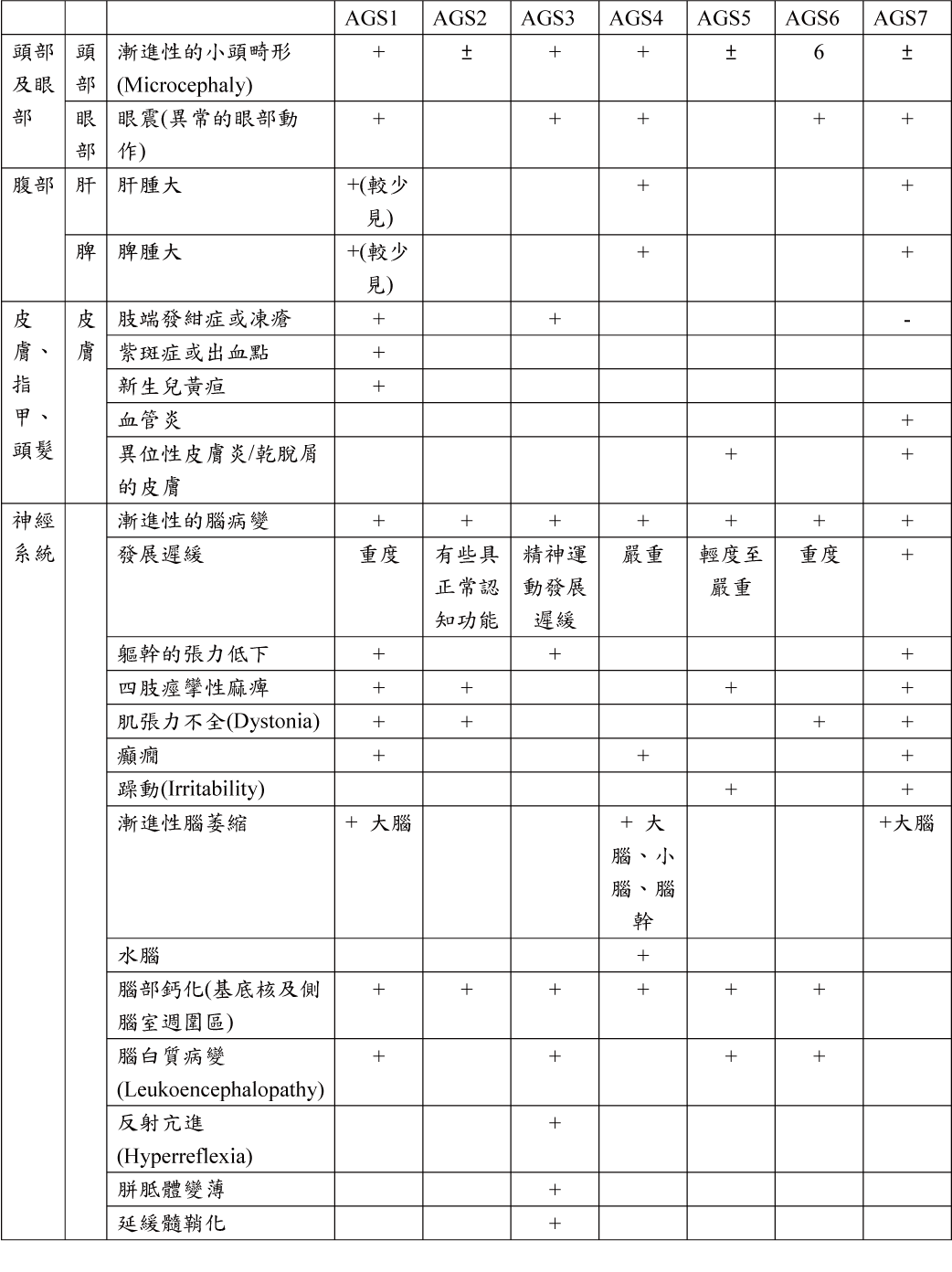
臨床表徵: AGS最典型的症狀是早發性腦病並伴隨顯著的智力和身體障礙,頭顱的CT和MRI的異常(基底神經節和白質鈣化)。
臨床症狀:
1. 腦病或/和智能障礙
2. 出生內一年出現小頭症
3. 肌張力不全、痙攣
4. 發燒(無感染)
5. 肝脾腫大
6. 手腳、耳朵等的皮膚出現凍瘡損傷、皮膚斑駁
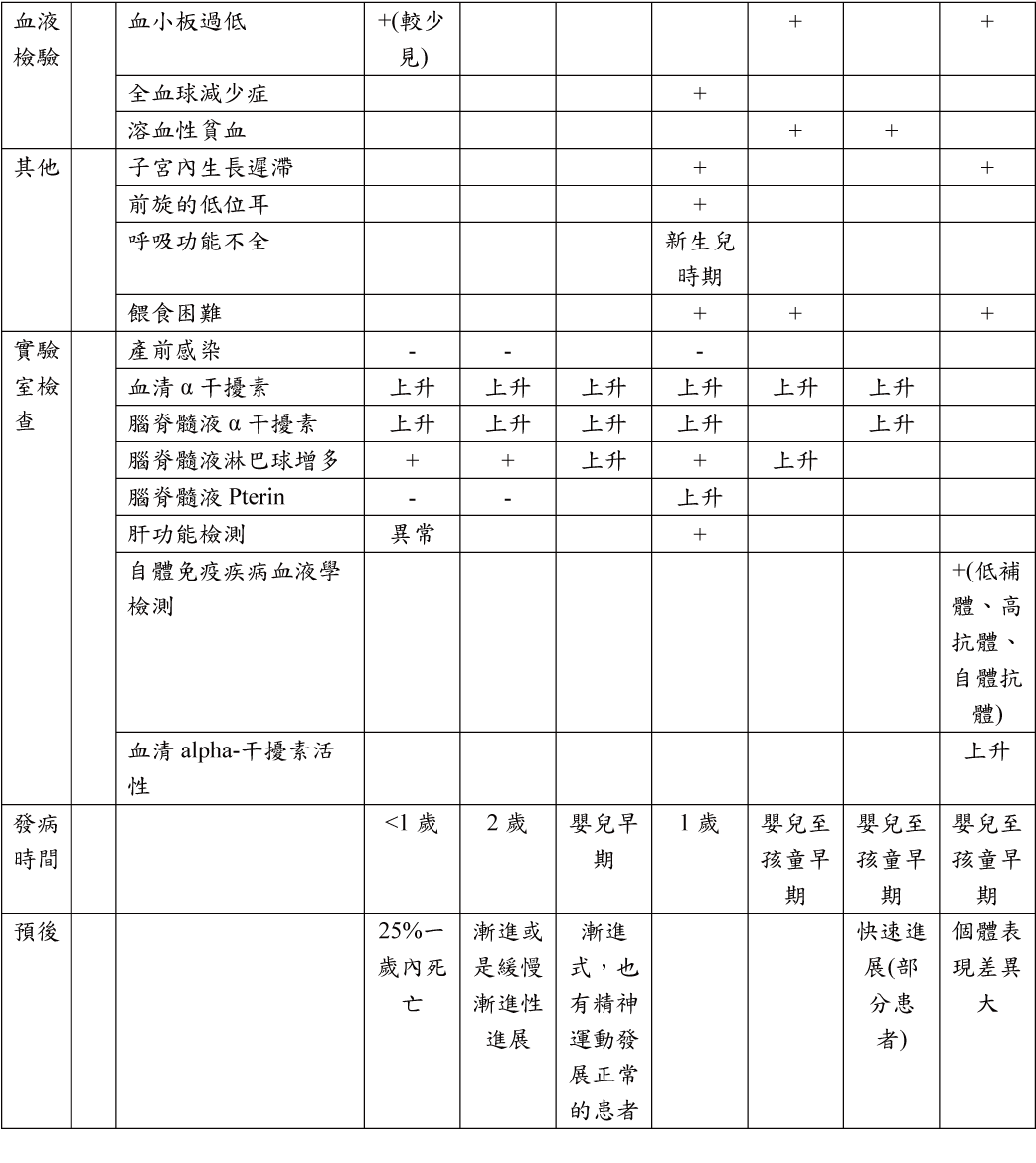
統整如下表:
診斷:
此症需要綜合臨床症狀、神經學影像及相關檢驗數值才可確認診斷,如頭部CT(基底節和白質的鈣化)及MRI(腦白質障礙)出現有明顯的異常,並在已知的相關基因中發現致病突變。約90%的AGS個案臨床表現和神經學結果一致性,並帶有相關基因突變。
(1)神經影像
鈣化:
CT掃描可看到顱內基底節和白質區域的鈣化,鈣化程度與神經功能預後的嚴重程度沒有相關性;在極少數情況下,無法看到鈣化的現象,因此就算沒有出現鈣化也不能排除此症。
腦白質退化:
許多患者出現低密度腦白質。在MRI分析上,常在腦室周圍的出現低密度現象。在嚴重的早發性病例中,在額骨上的腦白質病變表現尤為顯著。值得注意的是,在MRI上並不一錠看的到顱內鈣化的現象。
腦萎縮:
腦室周圍白質及腦溝進行性萎縮是一種非常常見的特徵。
小腦萎縮和腦幹萎縮可能會比較明顯。
具有ADAR 致病變異的話可以觀察到雙側紋狀體壞死。
腦血管病變:
具有SAMHD1 致病變異的話可以觀察到顱內動脈狹窄和動脈瘤。
(2)生化血球檢驗項目:
AGS的生化檢查會利用腦脊髓液(CSF)中的的白血球、干擾素α(IFN-α)和新蝶呤(neopterin)濃度進行評估。但在疾病早期階段,這些檢查數值可能是正常的。
干擾素-α(IFN-α):
腦脊髓液中的IFN-α的濃度上升(正常:2 IU/mL)。
在疾病的早期階段表現最高,干擾素-α的腦脊液濃度會在第三至四年回歸正常,通常IFN-α的濃度在腦脊液中比在血液高,但也可能是正常數值。
在懷孕週期26週之胎兒其血液中IFN-α就會有上升的情形。
腦脊液淋巴細胞增多:
淋巴細胞增多症的定義在腦脊隨液中超過5 lymphocytes/mm3,範圍從5到100 lymphocytes/mm3都有可能,雖然細胞增多的現象可能會持續數年,但隨著時間淋巴細胞的數量會遂漸減少。但是患者也可能沒有淋巴細胞增加的現象。
腦脊髓液中新蝶呤濃度(CSF neopterin):
疾病早期階段,新蝶呤的濃度表現最高,隨著時間的推移其可以恢復正常。而神經傳導代謝產物,如5HIAA、HVA和5MTHF的表現則是正常的。
排除AGS的鑑別診斷包括以下內容:
1、產前/出生前後期感染的證據,包括巨細胞病毒,弓形蟲,德國麻疹,單純皰疹病毒,和愛滋病毒。
2、已知的其他新陳代謝失調或神經退化性疾病。
(3)分子遺傳學檢測:
下列相關致病基因變異會引起AGS
Aicardi-Goutieres症候群致病基因及檢驗相關訊息如下:
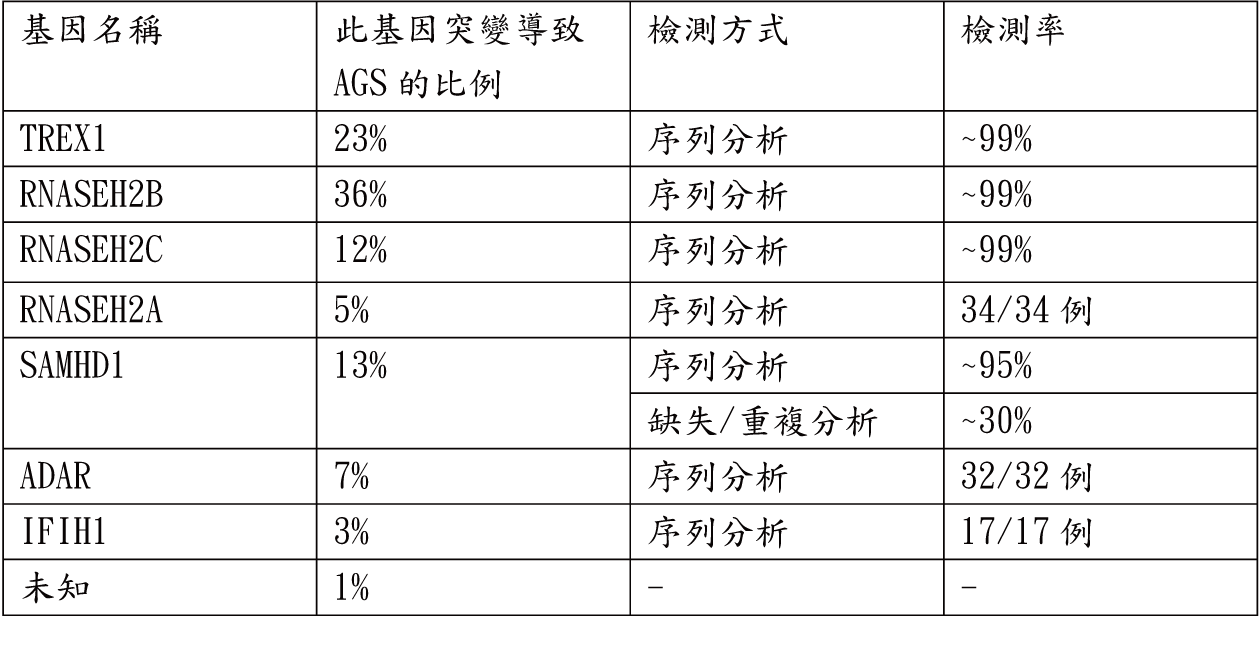
*除了SAMHD1基因在部分猶太人族群(方舟效應)中發現有片段缺失的現象外,其他基因並無缺失/重複分析的統計值。
約有8成的患嬰在懷孕、生產、新生兒時期都是正常的,除了TREX1突變的患者約有2成可在產前就發現腦部的鈣化,並在出生時即有神經學症狀、肝脾腫大、肝指數上升、血小板低下、類似先天性感染的症狀。其他大多數的嬰兒都在出生幾周內才開始出現症狀,腦病變的病程通常持續幾個月。帶有RNASEH2B突變的患者發病時間通常較晚(12個月大之後),並有較低的孩童時期死亡率及較好的智力。
治療:
胸腔物理治療和藥物來治療呼吸道併發症;注意飲食和餵食方式,確保足夠的熱量攝取,並避免呼吸道吸入異物;治療癲癇。
需定期監測新生兒時期有無糖尿病的表徵,幼兒時期持續的做眼科檢查,以評估是否有青光眼的情形;監測是否有脊柱側彎、胰島素依賴型糖尿病(IDDM)和甲狀腺功能低下的情況。
患者的死亡原因大多是因為疾病初期的腦損傷造成的繼發性問題所致。曾有報導指出患者活到30幾歲,且其疾病症狀並無明顯的惡化。
參考資料:
1. http://www.ncbi.nlm.nih.gov/books/NBK1475/
2. http://omim.org/phenotypicSeries/PS225750
編譯:余靜如
審稿:李妮鍾醫師 2015.02.03
2017年委託台大醫院基因醫學部 審稿
以上資料轉錄自「罕見遺傳疾病一點通」網頁 |