| 分類代碼:0911 |
| 疾病類別:09 |
| 疾病名稱: Freeman-Sheldon 氏症候群 ( Freeman-Sheldon Syndrome ) |
| 現階段政府公告之罕見疾病: 有 |
| 是否已發行該疾病之宣導單張:沒有 |
|
ICD-9-CM診斷代碼:272.7 ICD-10-CM診斷代碼:E 病因學: 此症又名Whistling Face Syndrome、Craniocarpotarsal Dysplasia或是Windmill-Vane-Hand syndrome。1938年Freeman 及 Sheldon首次描述病人有不正常的臉部特徵及骨骼異常。此項疾病屬於遠端關節彎曲型第二型 (distal arthrogryposis type 2, DA2A)。致病原因為MYH3基因缺陷所導致,可能與非進行性或緩慢進行性肌肉病變有關,其特徵為臉部、四肢以及呼吸肌肉纖維的先天分佈不均、骨骼異常。 Freeman-Sheldon氏症候群為位於染色體17短臂 (17p13.1),胚胎肌球蛋白重鏈 (embryonic myosin heavy chain, MYH3) 基因突變而導致疾病的發生。
此症屬罕見疾病,男女發生率相同。自從1938年Freeman及Sheldon博士首次提出討論此疾病以來,目前醫學文獻中已有超過100件案例被報告。 Freeman-Sheldon氏症候群屬於多重先天性攣縮症 (multiple congenital contractures, MCCs) 相關疾病群的一類,多重先天性攣縮症的發生率約為3000分之一。由於此疾病與其他先天型孿縮症往往具有相似的的病徵,因此有時會造成此疾病的誤診,造成此疾病的真實發生頻率往往相當的難估算。 遺傳模式: 此症大多為偶發,屬體染色體顯性遺傳。但偶有體染色體隱性模式被報告。若為顯性遺傳,個案其異常的基因可能來自於父母親其中一方或可能為個體新產生的基因突變。不論孩子性別,具有顯性遺傳性疾病的父母親皆有50%的機率將異常的基因傳給下一代。 遺傳表徵: 臨床特徵可分為三大部分: (1) 小下巴合併噘嘴(microstomia with pouting lips)
(2) 偏向小指的彎指畸形 (camptodactyly with ulnar deviation of fingers)
(3) 馬蹄形內翻足(talipes equinovarus)。
這類病人臉部的特徵是像帶面具般的臉 (mask face) 加上小嘴巴(microstomia)。病人還會有高拱的硬顎、小舌頭、長人中 (long philtrum) 以及特有的下巴H型凹陷。病人的牙齒也會發育不及咬合不正。有時候還有眼距過寬 (hypertelorism)、深眼廓 (deep-set eyes)、內貲贅皮 (epicanthal folds)、小眼裂 (narrow palpebral fissures)、鼻子很小、鼻翼發育不良且合併先天缺損等現象。 骨骼方面的異常包括四肢屈曲臠縮 (flexion contracture),風車葉板位置的手 (windmill vane position of hands),手偏向尺骨側 (ulnar deviation of hands),2-5指掌骨與指骨間的關節癵縮,大拇指內縮 (adducted thumb),但由於臨床嚴重度的不同以及表現型的差異,50% 的病人可能只有臉部有吹口哨的表情而手部是正常的。此外,病人也會合併隱睪及腹股溝疝氣。
除了顏面及骨骼異常外,也有案例有合併腦部發育異常以及神經性聽力異常。 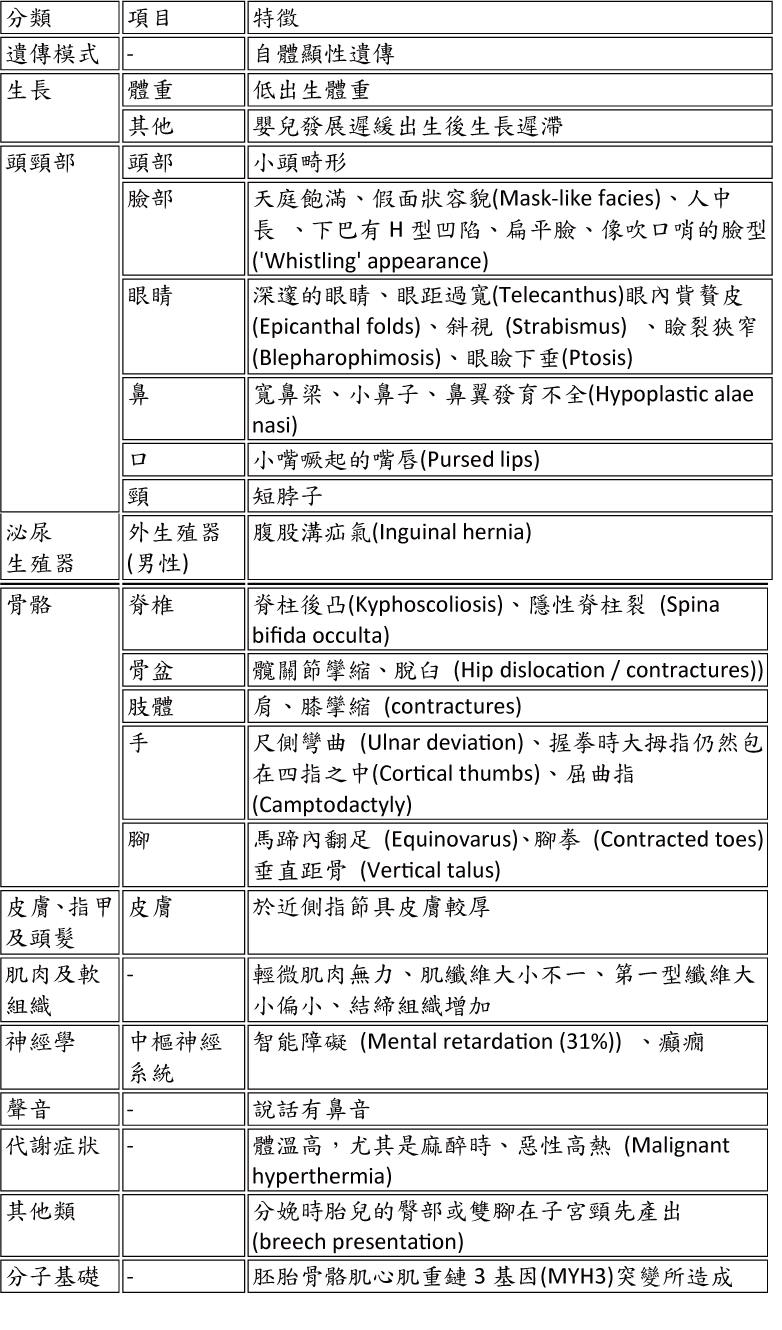 診斷: Freeman-Sheldon氏症候群的確診可根據完整的臨床評估、身體表徵的辨別、詳細的病史及專門的檢驗如影像學檢查而達成。在某些案例中,可藉由胎兒超音波找出異常的身體特徵而於出生前被診斷出Freeman-Sheldon氏症候群。 大部份患者的身體異常於出生時可能就已經特別的明顯了。然而每位患者的特徵及表現的差異相當大,且臉部特徵、手及腳的變異並不會在所有的個案中顯現出來。
電腦斷層掃描可用來偵測患者頭臉部及牙齒的異常特徵與其嚴重程度。X光也可顯示出顱骨後側的凹陷。此外,肌電圖可顯示手、腳及上顎的某些肌肉發育不全現象。
在一些案例中,使用手術或是顯微鏡肌肉組織切片的檢查可顯現出患者異常之纖維結締組織堆積。
治療: Freeman-Sheldon氏症候群的治療會針對每位患者所表現的各種症狀,直接針對症狀作治療。治療可能需要協同各專業團隊包括小兒科醫師、骨科醫師、牙科醫師、語言治療師、神經肌肉疾病專科醫師及其他健康照護專業人員提供患童全面性的治療計畫。 小口畸形通常會藉由手術或藉由每天數小時使用特別的裝置來擴大患者的嘴部。有些患者因為嘴巴太小而無法進食,需要矯正牙齒相關異常,如咬合不正的齒顎矯正治療,因此小口畸形的治療對於患者是必須的。
吞嚥困難可藉由一些裝置來維持頸部張力 (如:環形架頸牽引) 及矯正因枕骨後-壓迫性頸椎病變所引發的頸部肌肉僵硬以達改善效果。語言治療可改善患者語言及吞嚥相關舌頭的運動。
手術可用來矯正其他的身體異常,如: 足畸形 (club feet)、手部異常、顱面異常及膝蓋及手軸的攣縮現象。物理治療結合手術及支持性的措施可改善患者獨立地完成行走及執行其他動作。尺側偏斜 (Ulnar deviation) 通常會隨年齡增長而改善,不需要接受特別的矯正手術。
有些Freeman-Sheldon氏症的患者於接觸麻醉劑或肌肉鬆弛劑 (如:氟烷,halothane) 可能會引發似惡性高熱 (malignant hyperthermia-like) 反應。外科醫師、麻醉醫師、牙科醫師於執行手術時及使用麻醉劑時應將此項風險列入考量,開立特定處方藥時也必須考量患者的此項風險性。
早期介入治療相當的重要,可使Freeman-Sheldon氏症的患童減緩惡化程度。一些訓練如特殊教育、職能治療等,對患童也會有所助益。遺傳諮詢對於患者及其家庭會有所幫助。其他治療皆多屬症狀性及支持性的治療方式。
預後: 患者智力大多正常,生長發育可能會稍微遲滯,先天性心臟病的機率也不會上升,如無嚴重合併症,壽命與正常人無異。少部分病人有報告有嚴重的發展遲緩,這些人多是屬於體染色體隱性遺傳者,這類病人容易早逝,這可能是由於腦幹也受到影響以及進行性呼吸及餵食困難所致。 併發症方面,這類病人最嚴重的表現是吞嚥困難以及肺部的問題。由於下巴小,病人會有餵食及吞嚥困難的症狀。嘴巴構造異常除了造成吞嚥困難,還會造成呼吸到阻塞以致呼吸暫停、語言困難、鼻音重以及張口困難等現象。
由於肌肉張力不足,病人也可能有脊柱後側彎 (kyphoscoliosis),並進而造成限制性肺病。針對麻醉,由於病人的靜脈注射及插管皆屬困難,而且惡性體溫過高 (malignant hyperthermia) 或手術後肺部併發症可能性上升,所以麻醉是具有風險性的。
1.OMIM. Freeman-Sheldon Syndrome. (http://omim.org/entry/193700)
2.Kenneth LJ, Marilyn CJ, Miguel DC. Freeman-Sheldon Syndrome, Smith’s Recognizable Patterns of Human Malformation; 8th Edition, pp. 300-301.
3.National Organization for Rare Disorders (NORD). (n.d.). Freeman-Sheldon Syndrome. (https://rarediseases.org/rare-diseases/freeman-sheldon-syndrome/)
4.Ferrari D, Bettuzzi C, Donzelli O. Freeman-Sheldon syndrome. A case report and review of the literature. Chir Organi Mov. 2008;92(2):127-131.
5.Poling MI, Morales Corado JA, Chamberlain RL. Findings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis. Syst Rev. 2017;6(1):46.
6.Aalam, M., & colleagues. (1972). Windmill vane-like finger deformities. Zeitschrift für Orthopädie und ihre Grenzgebiete, 110(3), 395-398.
翻譯:林恩如 審閱:李妮鍾醫師 2014.08.29 2017年委託台大醫院基因醫學部 審稿 2024年張聿民醫師 審閱更新 以上資料轉載自「罕見遺傳疾病一點通」網頁 |
|
115年公益勸募字號: 衛部救字第1141364459號 罕見之愛 溫暖同行 |
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|